

De novo activating mutations drive clonal evolution and enhance clonal fitness in KMT2A-rearranged leukemia
- PMID: 29720585
- PMCID: PMC5932012
- DOI: 10.1038/s41467-018-04180-1
De novo activating mutations drive clonal evolution and enhance clonal fitness in KMT2A-rearranged leukemia
Abstract
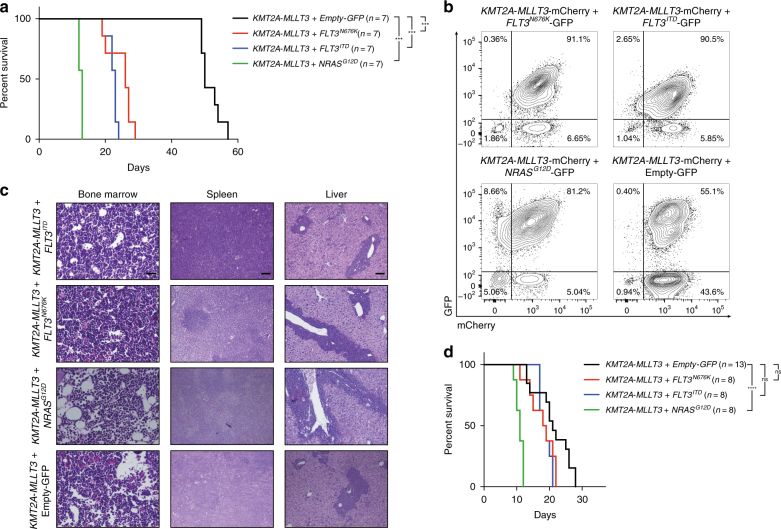
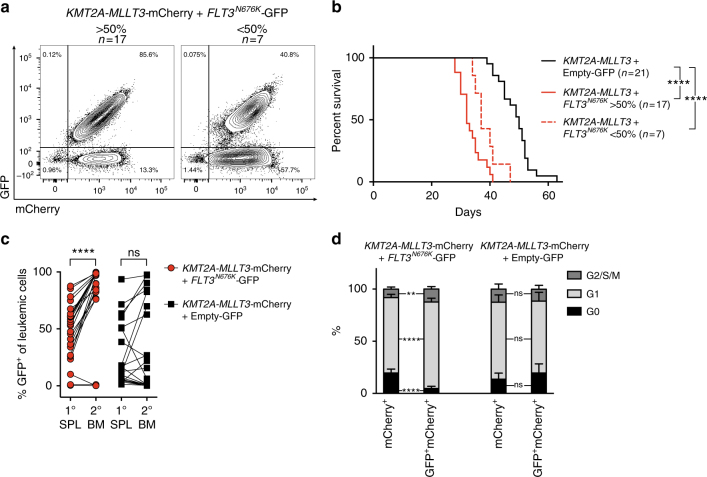
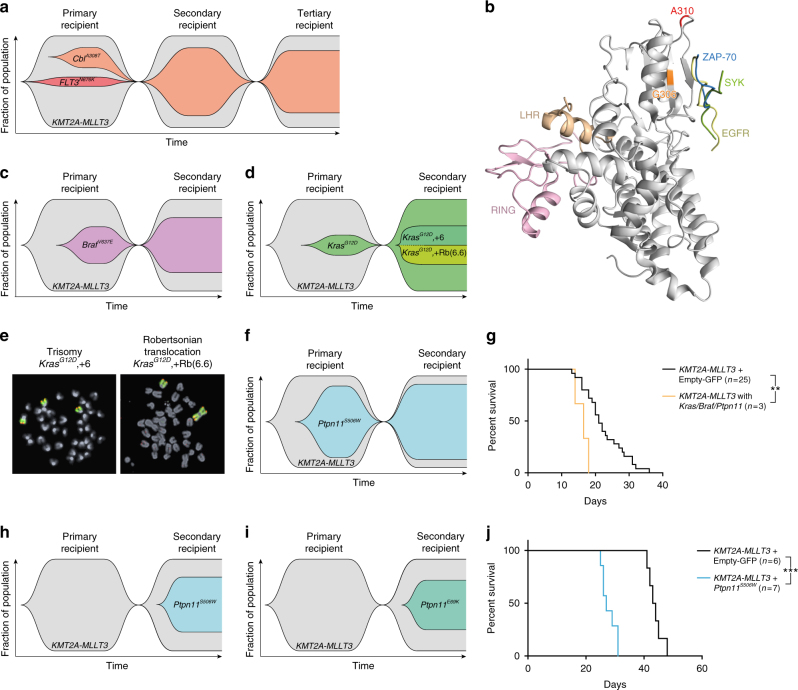
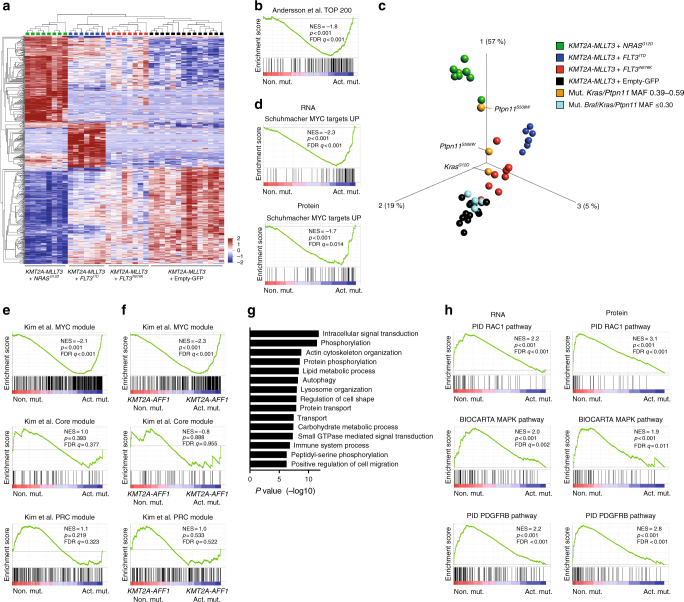
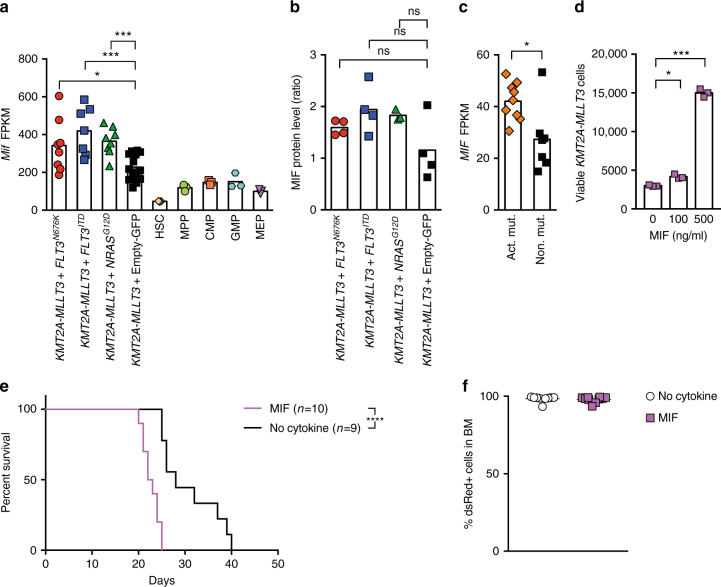
Activating signaling mutations are common in acute leukemia with KMT2A (previously MLL) rearrangements (KMT2A-R). These mutations are often subclonal and their biological impact remains unclear. Using a retroviral acute myeloid mouse leukemia model, we demonstrate that FLT3 ITD , FLT3 N676K , and NRAS G12D accelerate KMT2A-MLLT3 leukemia onset. Further, also subclonal FLT3 N676K mutations accelerate disease, possibly by providing stimulatory factors. Herein, we show that one such factor, MIF, promotes survival of mouse KMT2A-MLLT3 leukemia initiating cells. We identify acquired de novo mutations in Braf, Cbl, Kras, and Ptpn11 in KMT2A-MLLT3 leukemia cells that favored clonal expansion. During clonal evolution, we observe serial genetic changes at the Kras G12D locus, consistent with a strong selective advantage of additional Kras G12D . KMT2A-MLLT3 leukemias with signaling mutations enforce Myc and Myb transcriptional modules. Our results provide new insight into the biology of KMT2A-R leukemia with subclonal signaling mutations and highlight the importance of activated signaling as a contributing driver.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
