

Molecular Findings in Families with an Initial Diagnose of Autosomal Dominant Retinitis Pigmentosa (adRP)
- PMID: 29721949
- PMCID: PMC7138351
- DOI: 10.1007/978-3-319-75402-4_29
Molecular Findings in Families with an Initial Diagnose of Autosomal Dominant Retinitis Pigmentosa (adRP)
Abstract
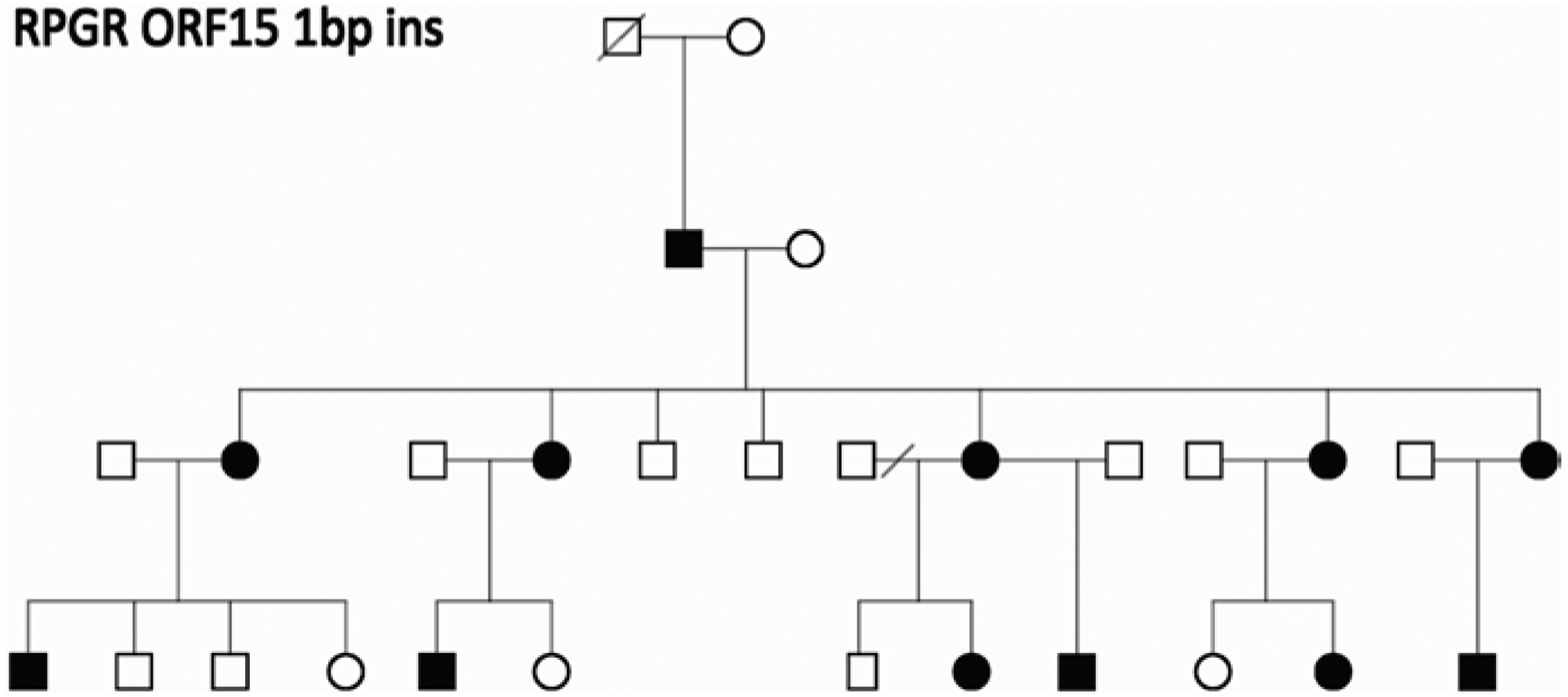
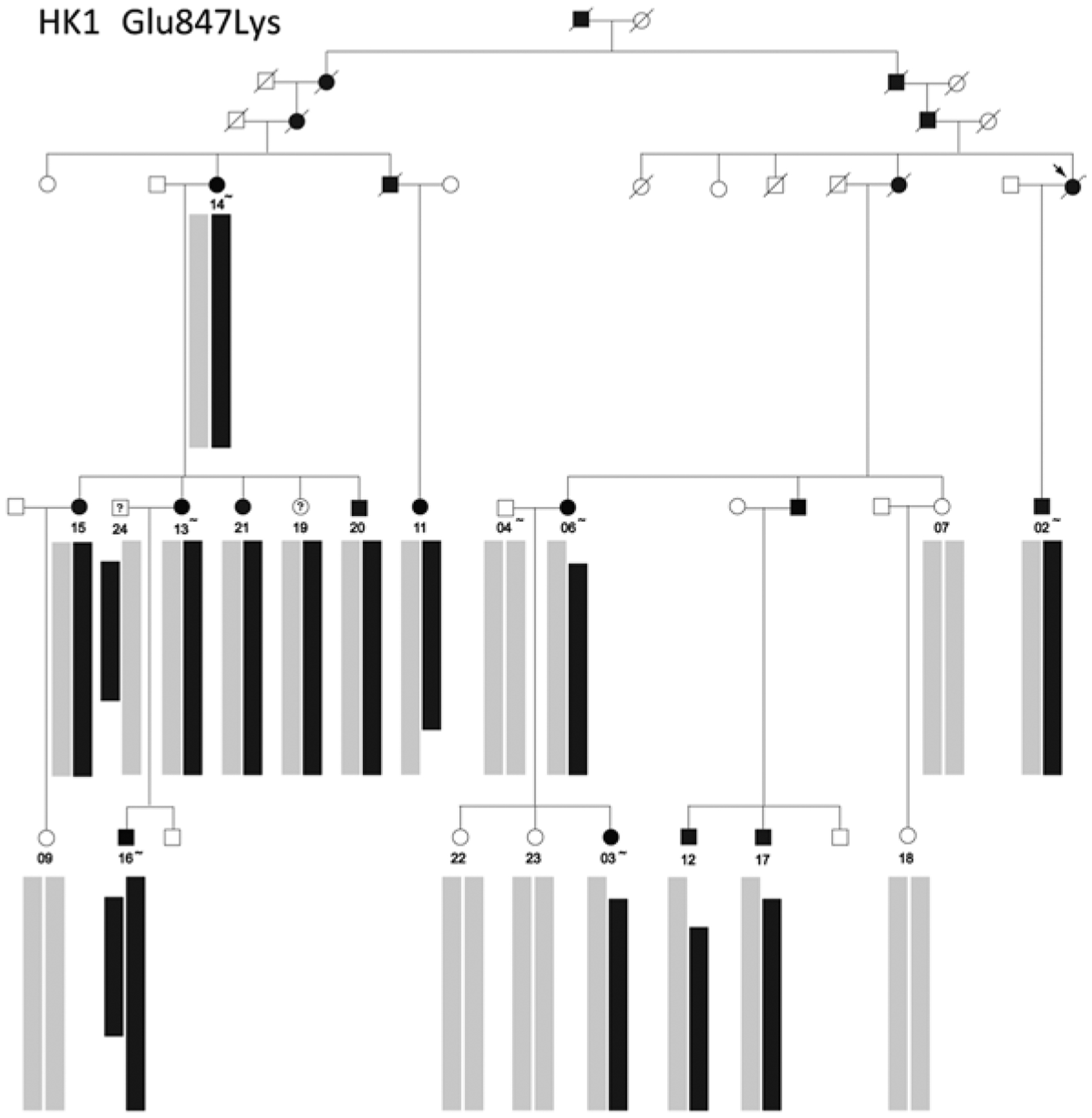
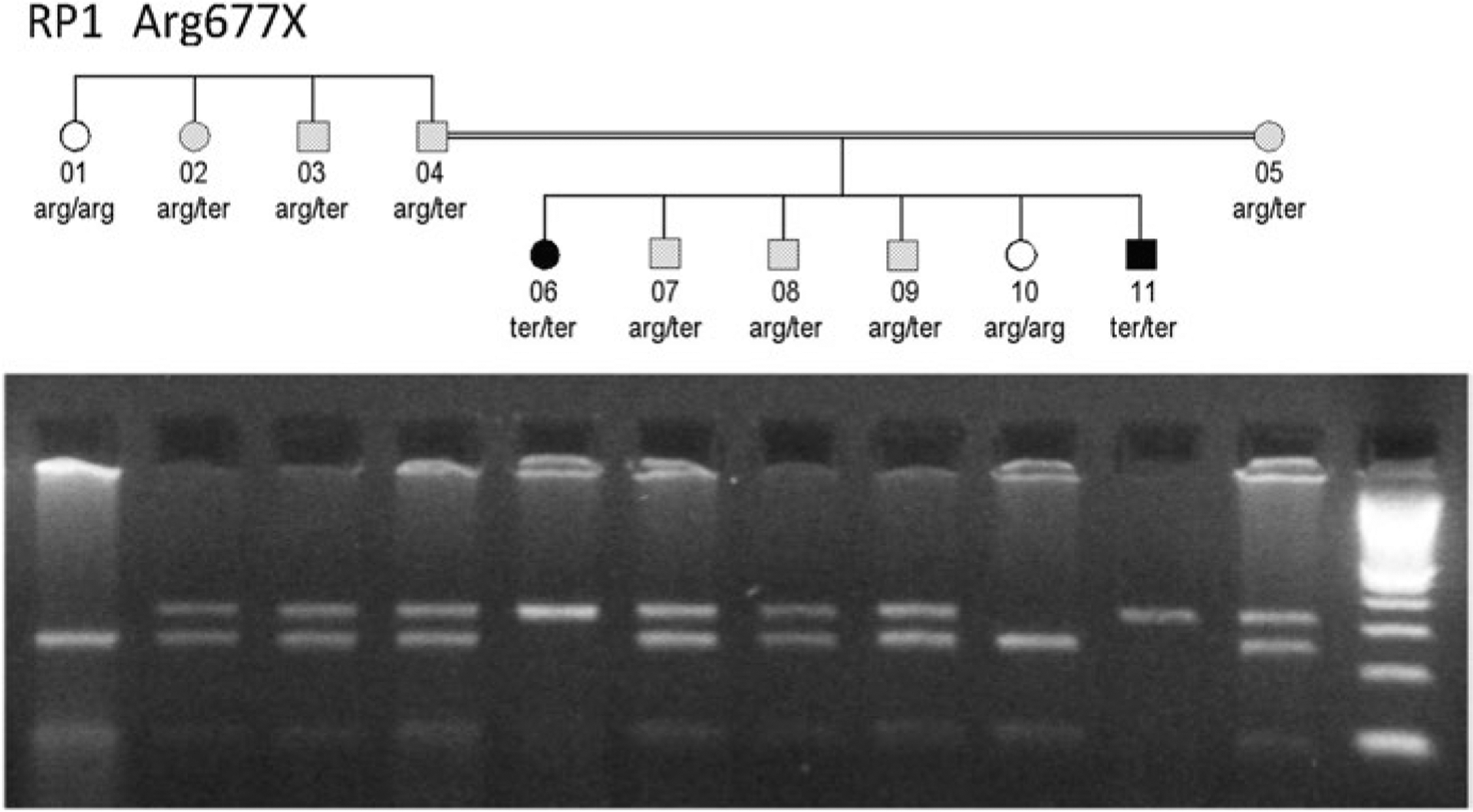
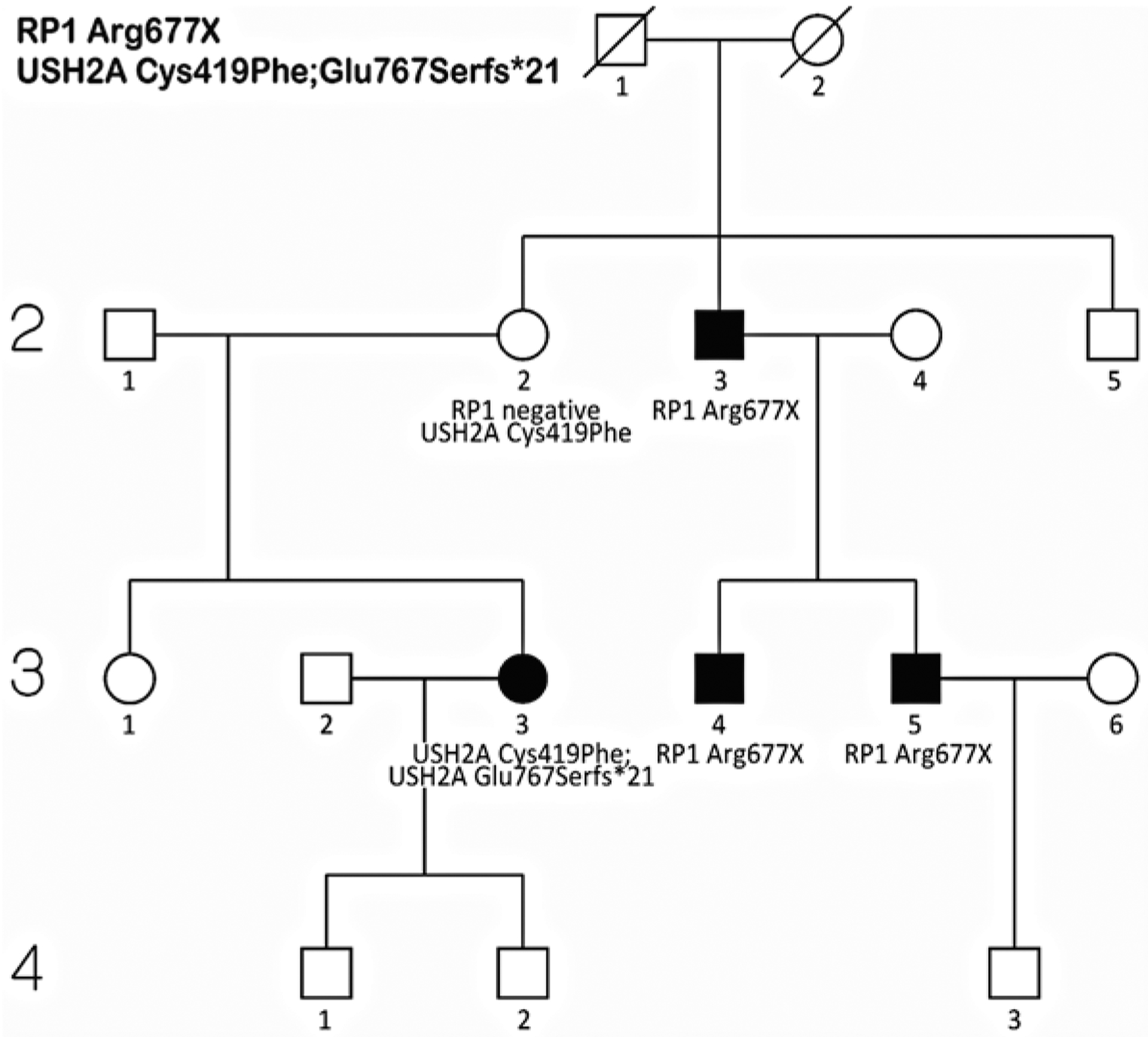
Genetic testing of probands in families with an initial diagnosis of autosomal dominant retinitis pigmentosa (adRP) usually confirms the diagnosis, but there are exceptions. We report results of genetic testing in a large cohort of adRP families with an emphasis on exceptional cases including X-linked RP with affected females; homozygous affected individuals in families with heterozygous, dominant disease; and independently segregating mutations in the same family. Genetic testing was conducted in more than 700 families with a provisional or probable diagnosis of adRP. Exceptions to the proposed mode of inheritance were extracted from our comprehensive patient and family database. In a subset of 300 well-characterized families with a probable diagnosis of adRP, 195 (70%) have dominant mutations in known adRP genes but 25 (8%) have X-linked mutations, 3 (1%) have multiple segregating mutations, and 3 (1%) have dominant-acting mutations in genes previously associated with recessive disease. It is currently possible to determine the underlying disease-causing gene and mutation in approximately 80% of families with an initial diagnosis of adRP, but 10% of "adRP" families have a variant mode of inheritance. Informed genetic diagnosis requires close collaboration between clinicians, genetic counselors, and laboratory scientists.
Keywords: Autosomal dominant retinitis pigmentosa; Linkage mapping; Next-generation sequencing; Prevalence of mutations; Retinitis pigmentosa (RP); Semidominant inheritance; X-linked retinitis pigmentosa.
Figures
References
-
- Bateson W (1908) The methods and scope of genetics: an inaugural lecture delivered 23 October 1908. Cambridge University Press, Fetter Lane, E.C, London, UK
-
- Berger W, Kloeckener-Gruissem B, Neidhardt J (2010) The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res 29:335–375 - PubMed
-
- Bowne SJ, Sullivan LS, Wheaton DK et al. (2015) Retinal targeted-capture next generation sequencing and CLIA confirmation in a representative range of patients with inherited retinal degeneration. Invest Ophthalmol Vis Sci E-Abstr 56:1241
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
