

Synaptic dysfunction in Alzheimer's disease: the effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention
- PMID: 29722304
- PMCID: PMC5950662
- DOI: 10.4103/1673-5374.230276
Synaptic dysfunction in Alzheimer's disease: the effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention
Abstract
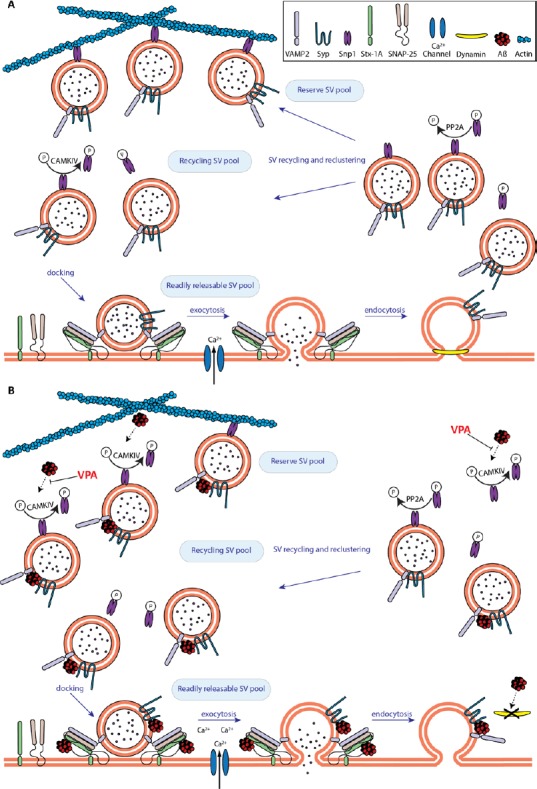
The most prevalent form of dementia in the elderly is Alzheimer's disease. A significant contributing factor to the progression of the disease appears to be the progressive accumulation of amyloid-β42 (Aβ42), a small hydrophobic peptide. Unfortunately, attempts to develop therapies targeting the accumulation of Aβ42 have not been successful to treat or even slow down the disease. It is possible that this failure is an indication that targeting downstream effects rather than the accumulation of the peptide itself might be a more effective approach. The accumulation of Aβ42 seems to affect various aspects of physiological cell functions. In this review, we provide an overview of the evidence that implicates Aβ42 in synaptic dysfunction, with a focus on how it contributes to defects in synaptic vesicle dynamics and neurotransmitter release. We discuss data that provide new insights on the Aβ42 induced pathology of Alzheimer's disease and a more detailed understanding of its contribution to the synaptic deficiencies that are associated with the early stages of the disease. Although the precise mechanisms that trigger synaptic dysfunction are still under investigation, the available data so far has enabled us to put forward a model that could be used as a guide to generate new therapeutic targets for pharmaceutical intervention.
Keywords: Alzheimer's disease; amyloid-β 42; neurotransmitter release; synaptic dysfunction; synaptic vesicles.
Conflict of interest statement
The authors declare no conflicts of interest
Figures
References
-
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–1576. - PubMed
-
- Amanatkar HR, Papagiannopoulos B, Grossberg GT. Analysis of recent failures of disease modifying therapies in Alzheimer's disease suggesting a new methodology for future studies. Expert Rev Neurother. 2017;17:7–16. - PubMed
-
- Arias C, Arrieta I, Tapia R. beta-Amyloid peptide fragment 25-35 potentiates the calcium-dependent release of excitatory amino acids from depolarized hippocampal slices. J Neurosci Res. 1995;41:561–566. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
