

bigSCale: an analytical framework for big-scale single-cell data
- PMID: 29724792
- PMCID: PMC5991513
- DOI: 10.1101/gr.230771.117
bigSCale: an analytical framework for big-scale single-cell data
Abstract
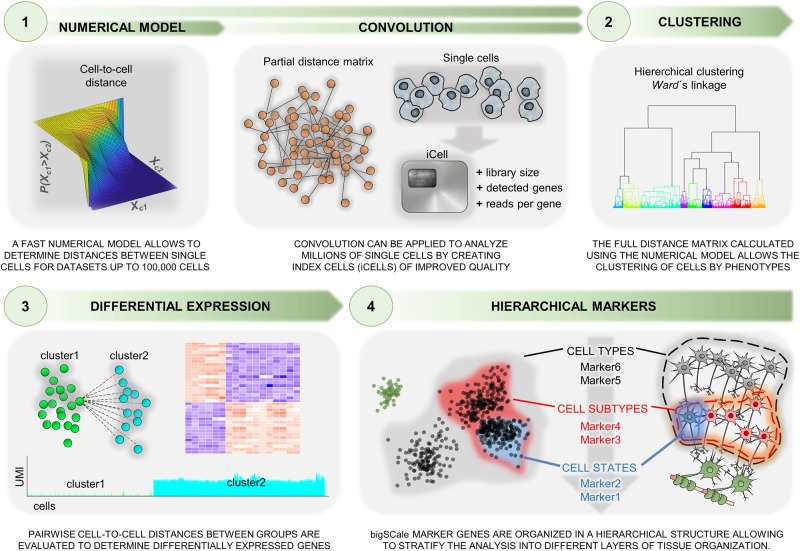
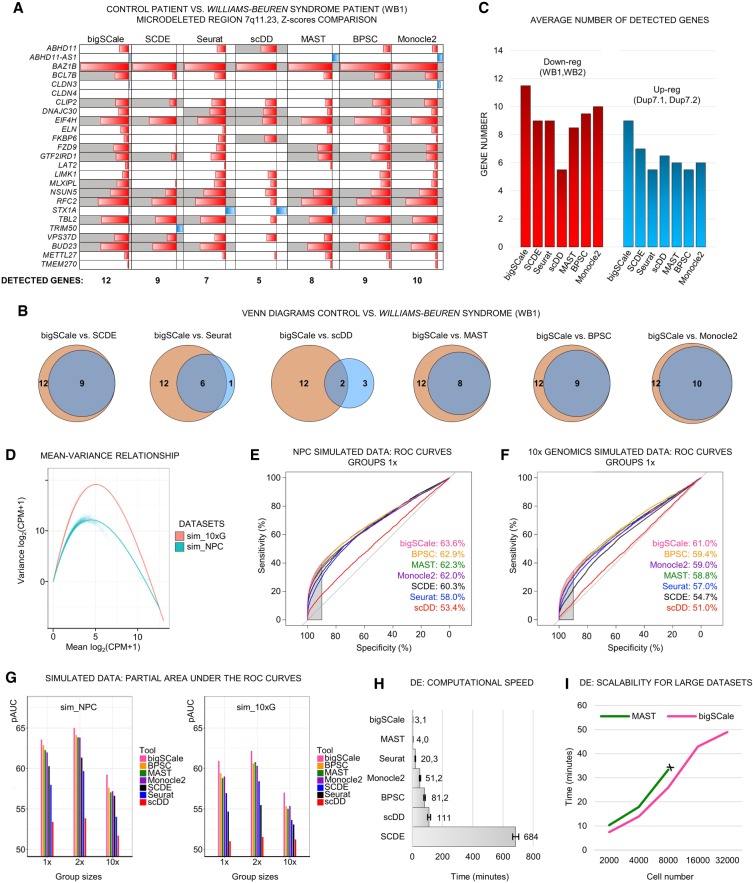
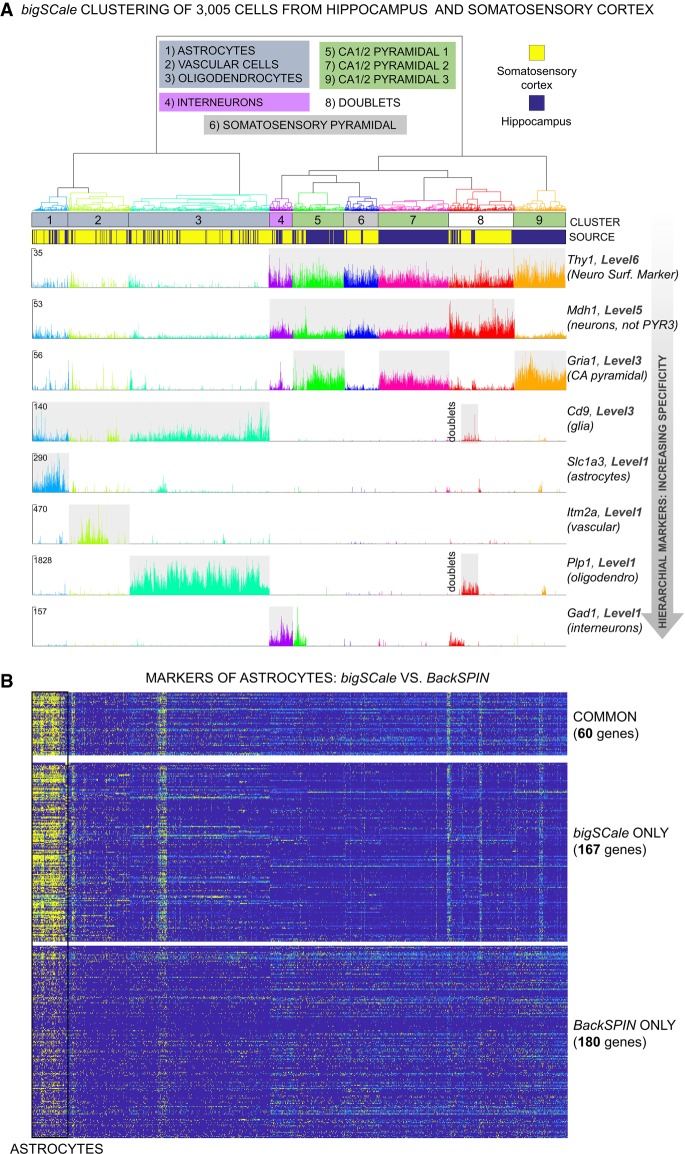
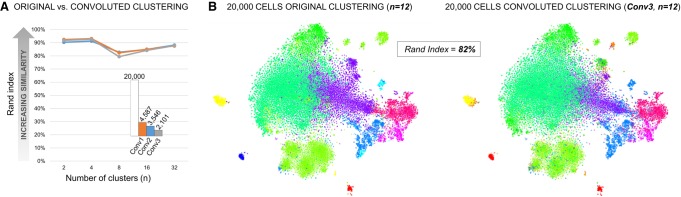
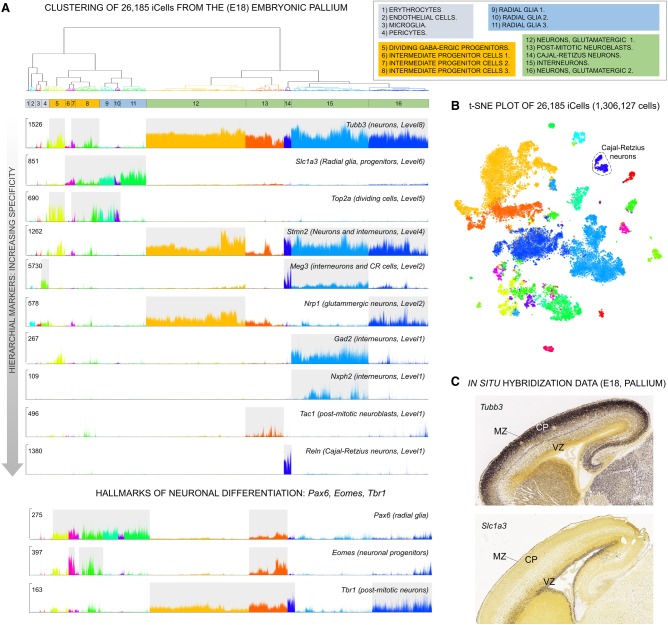
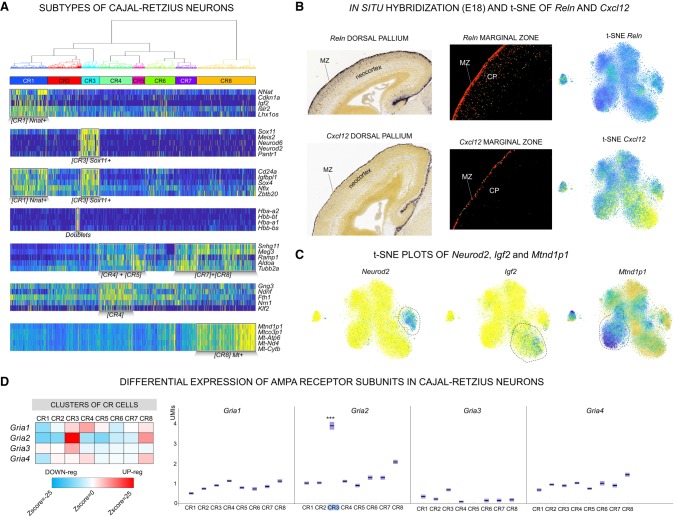
Single-cell RNA sequencing (scRNA-seq) has significantly deepened our insights into complex tissues, with the latest techniques capable of processing tens of thousands of cells simultaneously. Analyzing increasing numbers of cells, however, generates extremely large data sets, extending processing time and challenging computing resources. Current scRNA-seq analysis tools are not designed to interrogate large data sets and often lack sensitivity to identify marker genes. With bigSCale, we provide a scalable analytical framework to analyze millions of cells, which addresses the challenges associated with large data sets. To handle the noise and sparsity of scRNA-seq data, bigSCale uses large sample sizes to estimate an accurate numerical model of noise. The framework further includes modules for differential expression analysis, cell clustering, and marker identification. A directed convolution strategy allows processing of extremely large data sets, while preserving transcript information from individual cells. We evaluated the performance of bigSCale using both a biological model of aberrant gene expression in patient-derived neuronal progenitor cells and simulated data sets, which underlines the speed and accuracy in differential expression analysis. To test its applicability for large data sets, we applied bigSCale to assess 1.3 million cells from the mouse developing forebrain. Its directed down-sampling strategy accumulates information from single cells into index cell transcriptomes, thereby defining cellular clusters with improved resolution. Accordingly, index cell clusters identified rare populations, such as reelin (Reln)-positive Cajal-Retzius neurons, for which we report previously unrecognized heterogeneity associated with distinct differentiation stages, spatial organization, and cellular function. Together, bigSCale presents a solution to address future challenges of large single-cell data sets.
© 2018 Iacono et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Chauvin S, Sobel A. 2015. Neuronal stathmins: a family of phosphoproteins cooperating for neuronal development, plasticity and regeneration. Prog Neurobiol 126: 1–18. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases