

Copy number variant analysis using genome-wide mate-pair sequencing
- PMID: 29726617
- PMCID: PMC6117203
- DOI: 10.1002/gcc.5
Copy number variant analysis using genome-wide mate-pair sequencing
Abstract
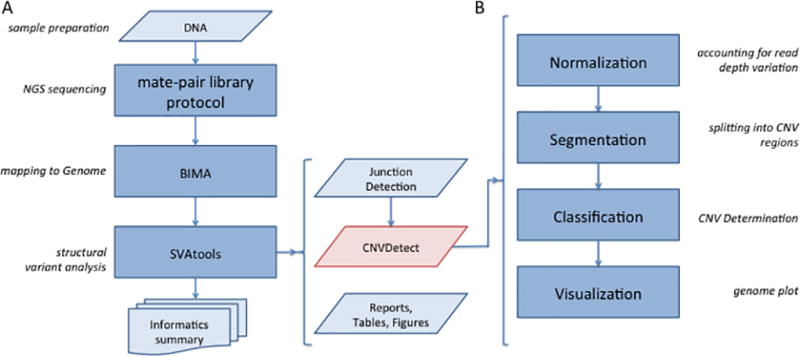
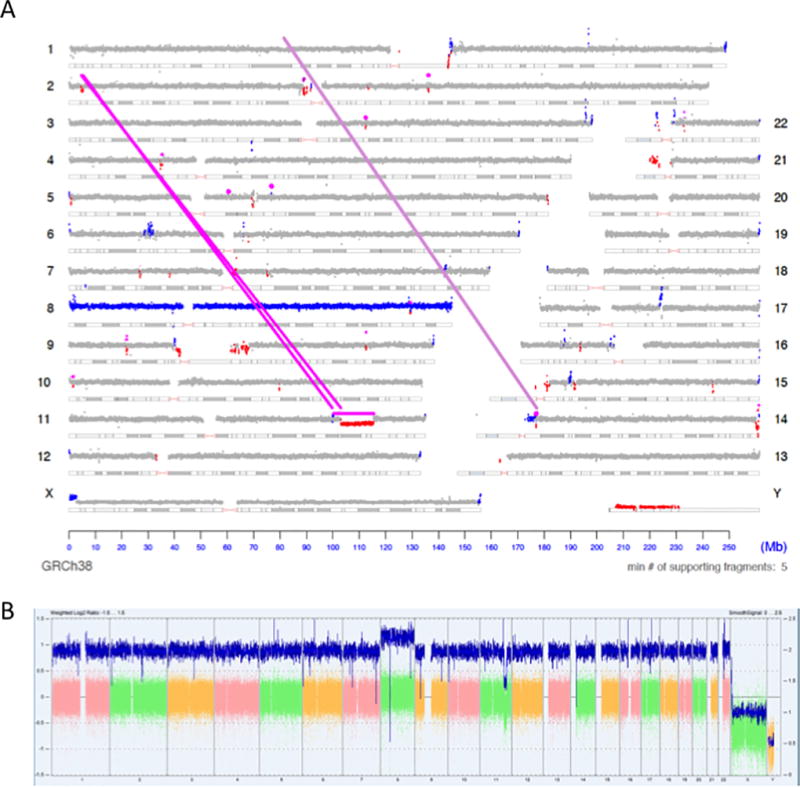
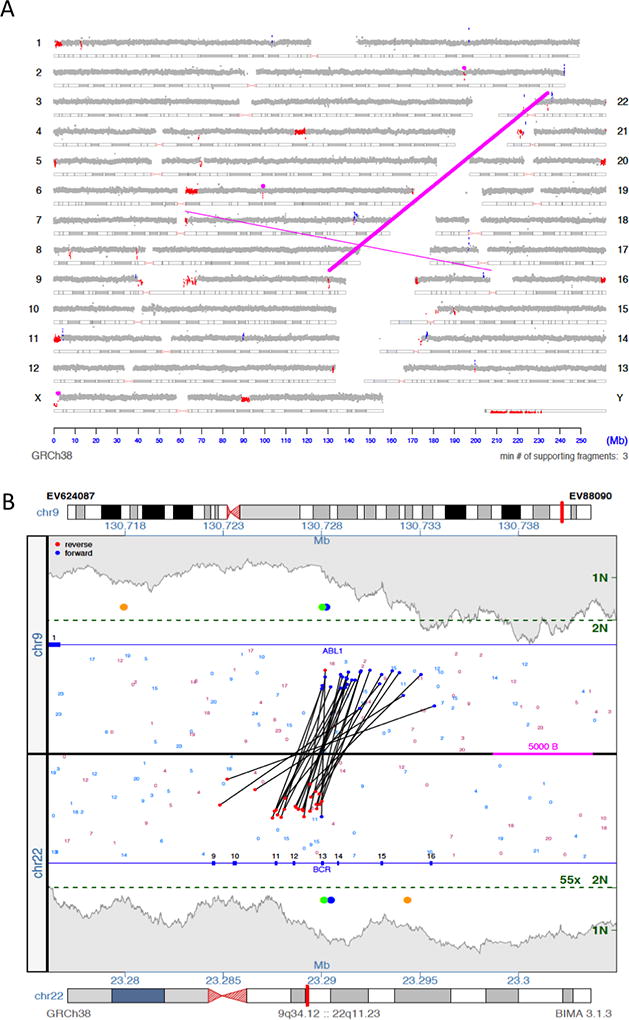
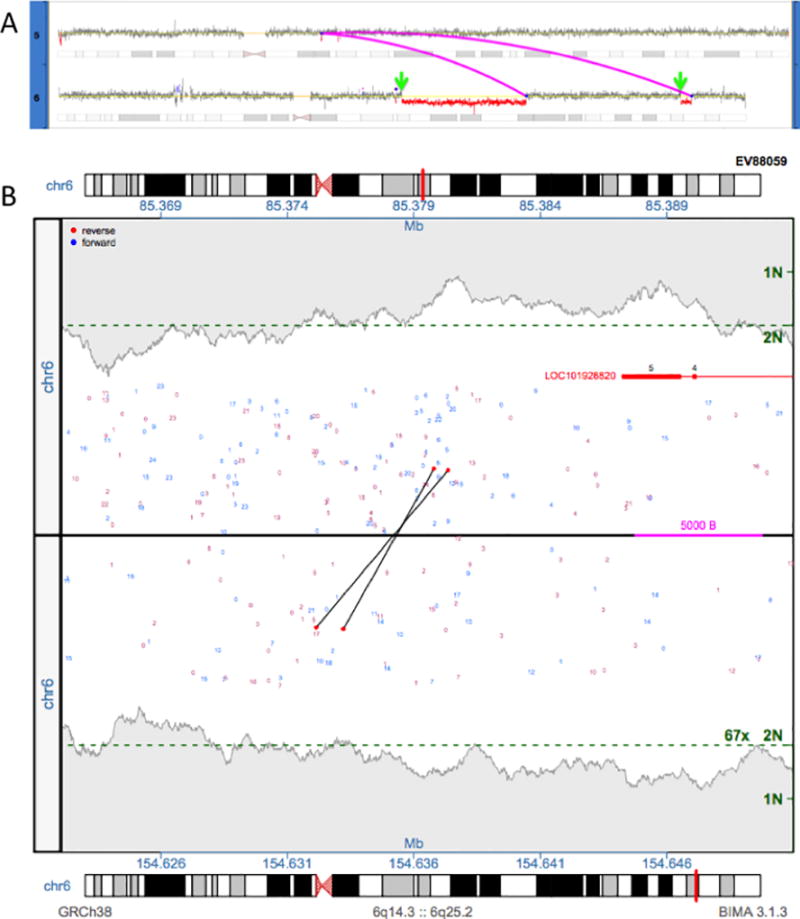
Copy number variation (CNV) is a common form of structural variation detected in human genomes, occurring as both constitutional and somatic events. Cytogenetic techniques like chromosomal microarray (CMA) are widely used in analyzing CNVs. However, CMA techniques cannot resolve the full nature of these structural variations (i.e. the orientation and location of associated breakpoint junctions) and must be combined with other cytogenetic techniques, such as karyotyping or FISH, to do so. This makes the development of a next-generation sequencing (NGS) approach capable of resolving both CNVs and breakpoint junctions desirable. Mate-pair sequencing (MPseq) is a NGS technology designed to find large structural rearrangements across the entire genome. Here we present an algorithm capable of performing copy number analysis from mate-pair sequencing data. The algorithm uses a step-wise procedure involving normalization, segmentation, and classification of the sequencing data. The segmentation technique combines both read depth and discordant mate-pair reads to increase the sensitivity and resolution of CNV calls. The method is particularly suited to MPseq, which is designed to detect breakpoint junctions at high resolution. This allows for the classification step to accurately calculate copy number levels at the relatively low read depth of MPseq. Here we compare results for a series of hematological cancer samples that were tested with CMA and MPseq. We demonstrate comparable sensitivity to the state-of-the-art CMA technology, with the benefit of improved breakpoint resolution. The algorithm provides a powerful analytical tool for the analysis of MPseq results in cancer.
Keywords: bioinformatics; cancer genetics; chromosomal rearrangements; copy number variant analysis; next-generation sequencing.
© 2018 Wiley Periodicals, Inc.
Figures
References
-
- Moncunill V, Gonzalez S, Bea S, et al. Comprehensive characterization of complex structural variations in cancer by directly comparing genome sequence reads. Nat Biotech. 2014;32(11):1106–1112. - PubMed
-
- Tubio JMC. Somatic structural variation and cancer. Briefings in Functional Genomics. 2015;14(5):339–351. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
