

Spliceosome Profiling Visualizes Operations of a Dynamic RNP at Nucleotide Resolution
- PMID: 29727661
- PMCID: PMC5940017
- DOI: 10.1016/j.cell.2018.03.020
Spliceosome Profiling Visualizes Operations of a Dynamic RNP at Nucleotide Resolution
Abstract
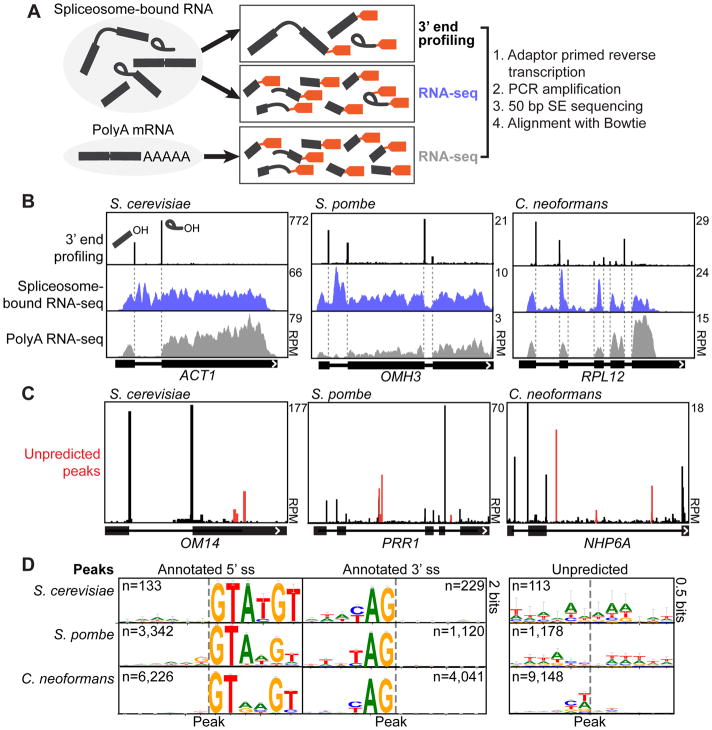
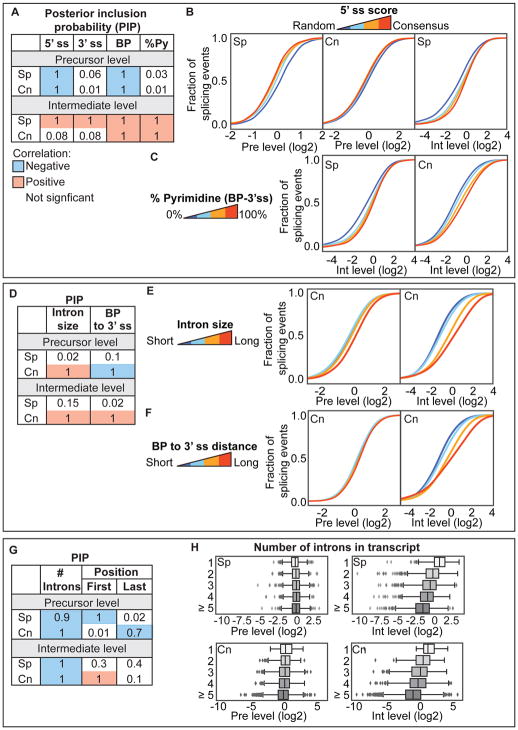
Tools to understand how the spliceosome functions in vivo have lagged behind advances in the structural biology of the spliceosome. Here, methods are described to globally profile spliceosome-bound pre-mRNA, intermediates, and spliced mRNA at nucleotide resolution. These tools are applied to three yeast species that span 600 million years of evolution. The sensitivity of the approach enables the detection of canonical and non-canonical events, including interrupted, recursive, and nested splicing. This application of statistical modeling uncovers independent roles for the size and position of the intron and the number of introns per transcript in substrate progression through the two catalytic stages. These include species-specific inputs suggestive of spliceosome-transcriptome coevolution. Further investigations reveal the ATP-dependent discard of numerous endogenous substrates after spliceosome assembly in vivo and connect this discard to intron retention, a form of splicing regulation. Spliceosome profiling is a quantitative, generalizable global technology used to investigate an RNP central to eukaryotic gene expression.
Keywords: pre-mRNA splicing; spliceosome; splicing catalysis; splicing fidelity.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures





Comment in
-
Scrutinizing spliceosomes.Nat Rev Genet. 2018 Jul;19(7):401. doi: 10.1038/s41576-018-0019-9. Nat Rev Genet. 2018. PMID: 29765162 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
