

Profiling and Leveraging Relatedness in a Precision Medicine Cohort of 92,455 Exomes
- PMID: 29727688
- PMCID: PMC5986700
- DOI: 10.1016/j.ajhg.2018.03.012
Profiling and Leveraging Relatedness in a Precision Medicine Cohort of 92,455 Exomes
Abstract
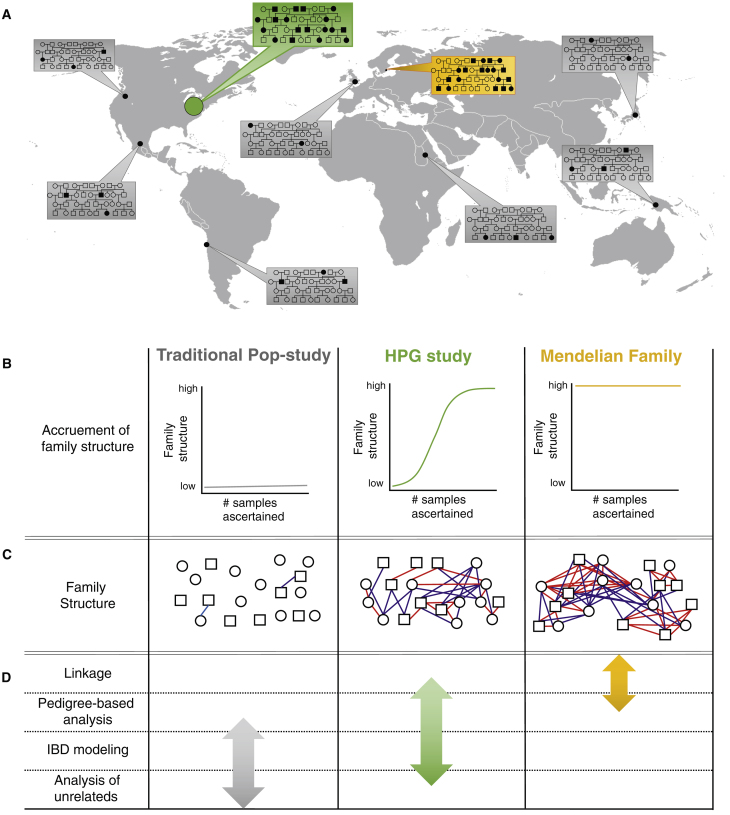
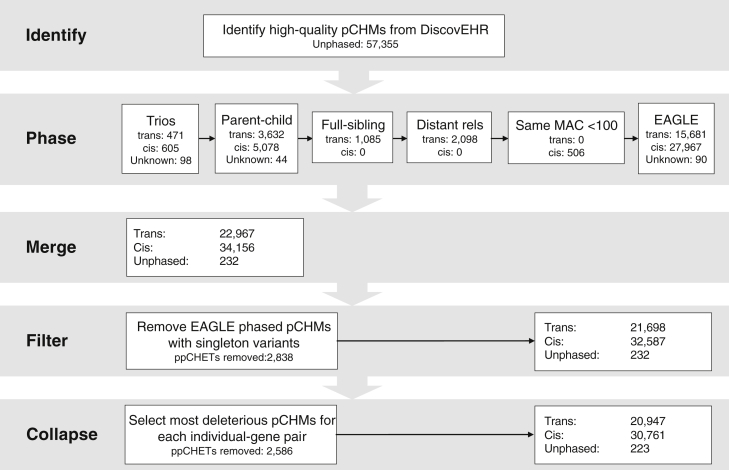
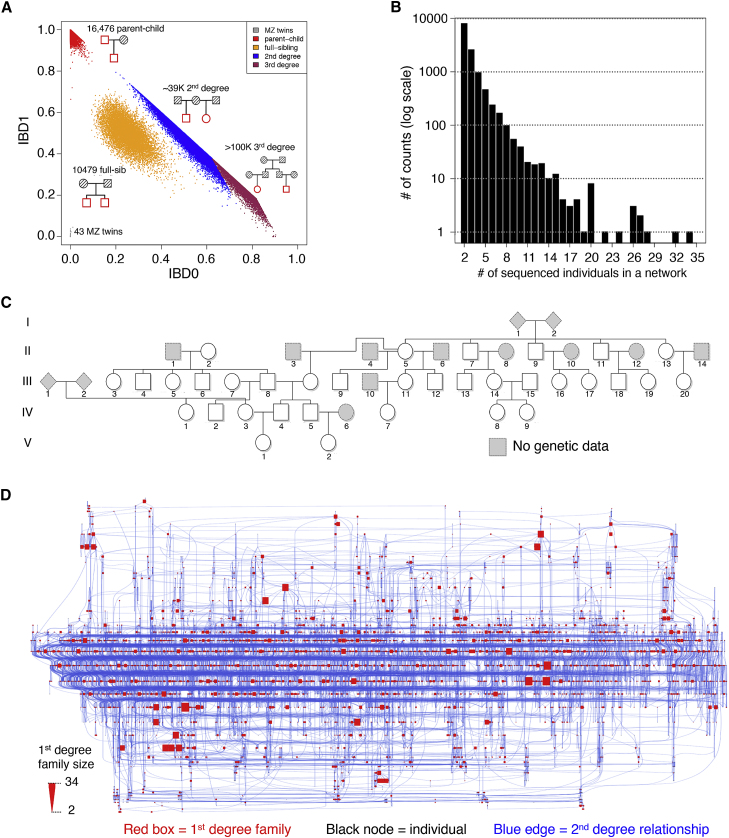
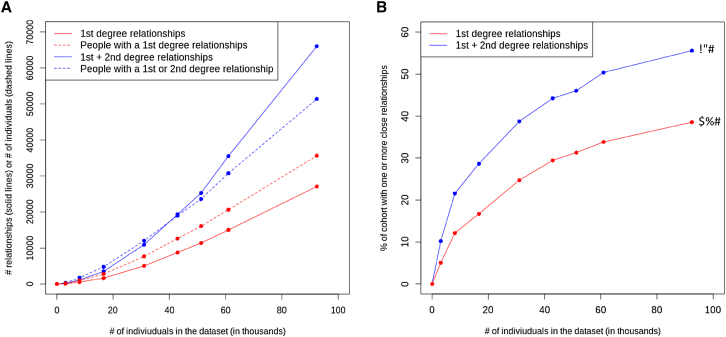
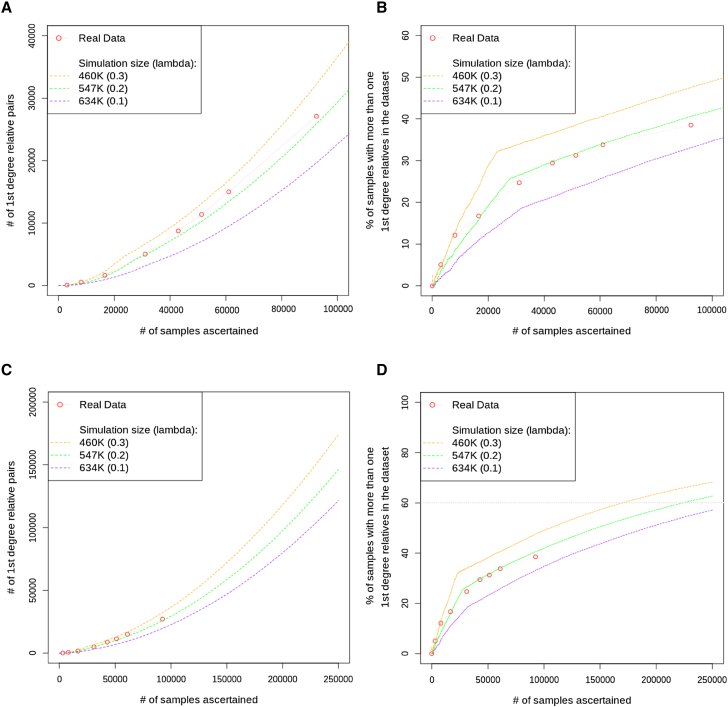
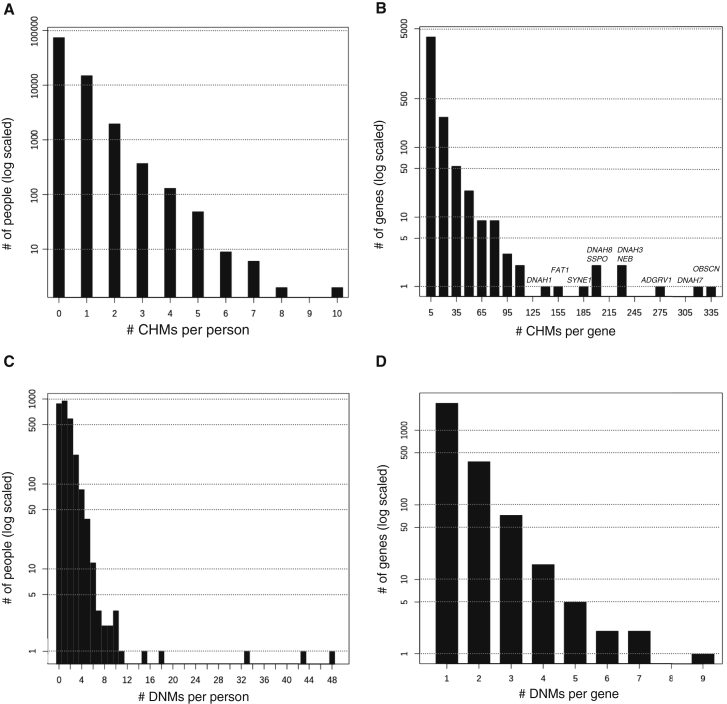
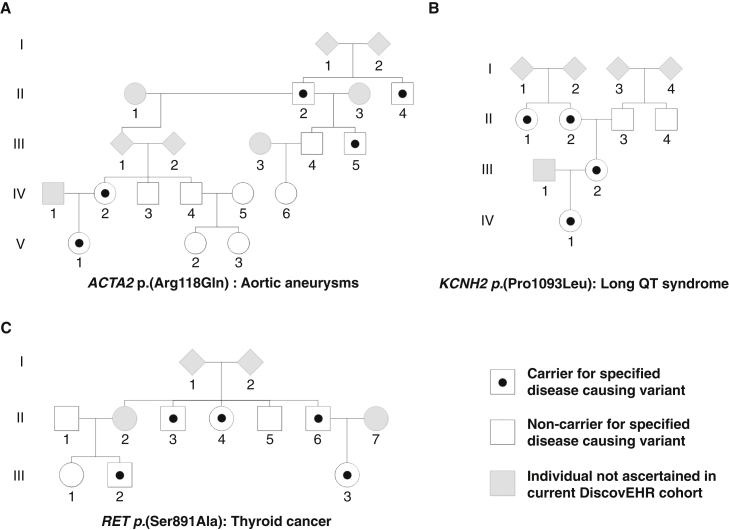
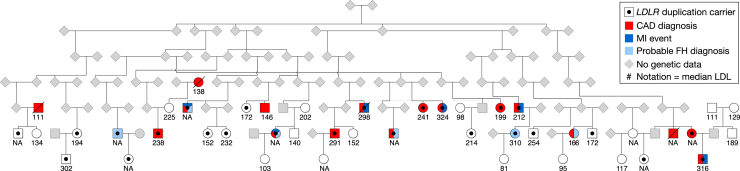
Large-scale human genetics studies are ascertaining increasing proportions of populations as they continue growing in both number and scale. As a result, the amount of cryptic relatedness within these study cohorts is growing rapidly and has significant implications on downstream analyses. We demonstrate this growth empirically among the first 92,455 exomes from the DiscovEHR cohort and, via a custom simulation framework we developed called SimProgeny, show that these measures are in line with expectations given the underlying population and ascertainment approach. For example, within DiscovEHR we identified ∼66,000 close (first- and second-degree) relationships, involving 55.6% of study participants. Our simulation results project that >70% of the cohort will be involved in these close relationships, given that DiscovEHR scales to 250,000 recruited individuals. We reconstructed 12,574 pedigrees by using these relationships (including 2,192 nuclear families) and leveraged them for multiple applications. The pedigrees substantially improved the phasing accuracy of 20,947 rare, deleterious compound heterozygous mutations. Reconstructed nuclear families were critical for identifying 3,415 de novo mutations in ∼1,783 genes. Finally, we demonstrate the segregation of known and suspected disease-causing mutations, including a tandem duplication that occurs in LDLR and causes familial hypercholesterolemia, through reconstructed pedigrees. In summary, this work highlights the prevalence of cryptic relatedness expected among large healthcare population-genomic studies and demonstrates several analyses that are uniquely enabled by large amounts of cryptic relatedness.
Keywords: compound heterozygous mutation phasing; cryptic relatedness; de novo mutations; exome sequencing; familial hypercholesterolemia; family structure; healthcare population-based genetic study; identity by decent; pedigree reconstruction; precision medicine; relationship inference.
Copyright © 2018 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Dewey F.E., Murray M.F., Overton J.D., Habegger L., Leader J.B., Fetterolf S.N., O’Dushlaine C., Van Hout C.V., Staples J., Gonzaga-Jauregui C. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354:aaf6814. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
