

FUN-LDA: A Latent Dirichlet Allocation Model for Predicting Tissue-Specific Functional Effects of Noncoding Variation: Methods and Applications
- PMID: 29727691
- PMCID: PMC5986983
- DOI: 10.1016/j.ajhg.2018.03.026
FUN-LDA: A Latent Dirichlet Allocation Model for Predicting Tissue-Specific Functional Effects of Noncoding Variation: Methods and Applications
Abstract
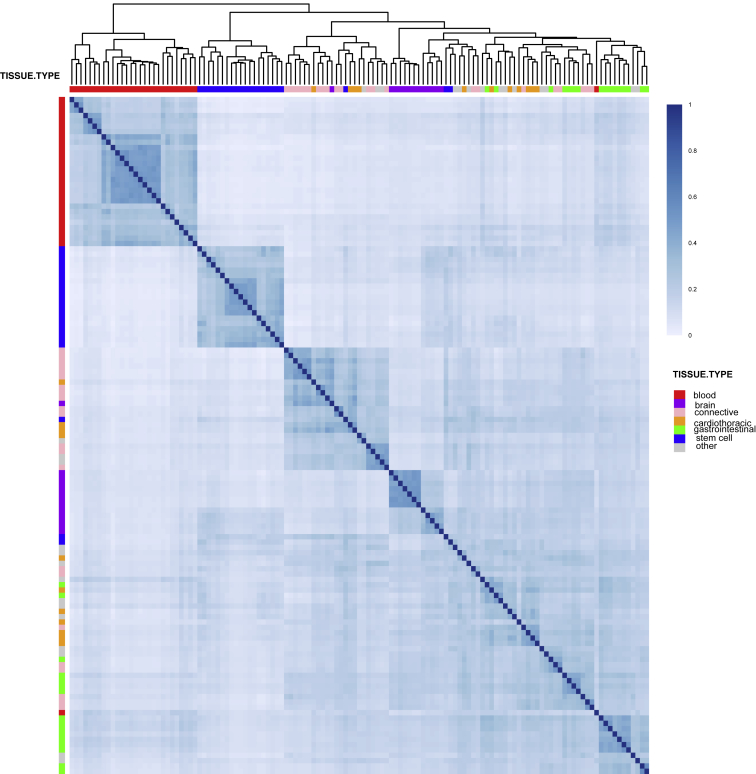
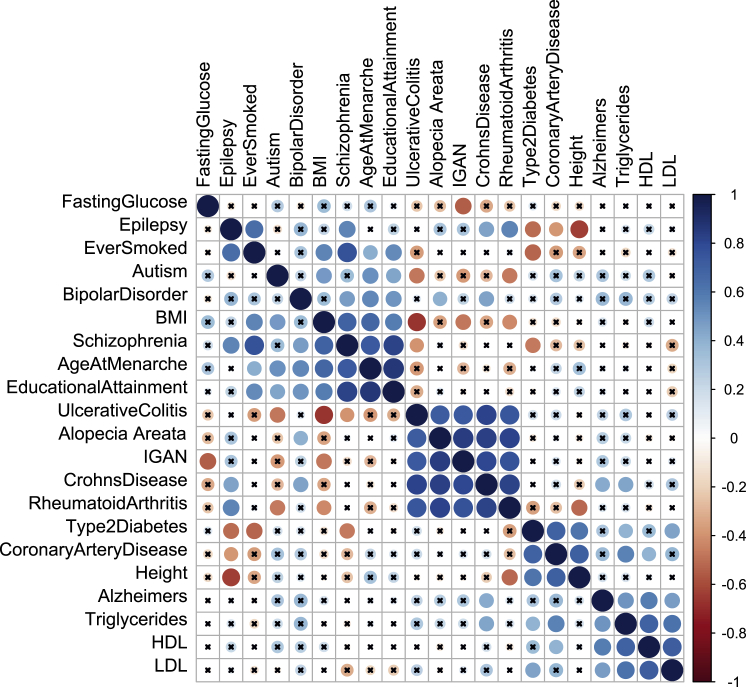
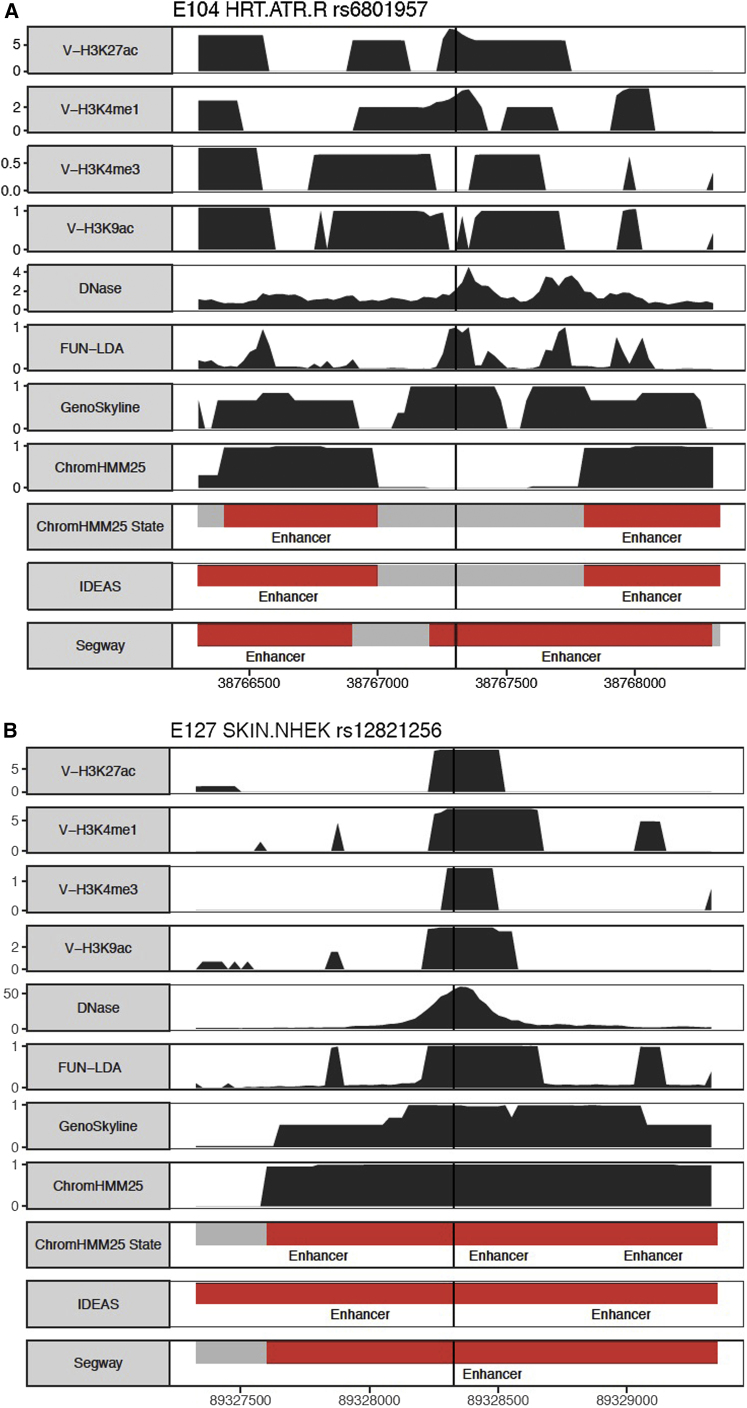
We describe a method based on a latent Dirichlet allocation model for predicting functional effects of noncoding genetic variants in a cell-type- and/or tissue-specific way (FUN-LDA). Using this unsupervised approach, we predict tissue-specific functional effects for every position in the human genome in 127 different tissues and cell types. We demonstrate the usefulness of our predictions by using several validation experiments. Using eQTL data from several sources, including the GTEx project, Geuvadis project, and TwinsUK cohort, we show that eQTLs in specific tissues tend to be most enriched among the predicted functional variants in relevant tissues in Roadmap. We further show how these integrated functional scores can be used for (1) deriving the most likely cell or tissue type causally implicated for a complex trait by using summary statistics from genome-wide association studies and (2) estimating a tissue-based correlation matrix of various complex traits. We found large enrichment of heritability in functional components of relevant tissues for various complex traits, and FUN-LDA yielded higher enrichment estimates than existing methods. Finally, using experimentally validated functional variants from the literature and variants possibly implicated in disease by previous studies, we rigorously compare FUN-LDA with state-of-the-art functional annotation methods and show that FUN-LDA has better prediction accuracy and higher resolution than these methods. In particular, our results suggest that tissue- and cell-type-specific functional prediction methods tend to have substantially better prediction accuracy than organism-level prediction methods. Scores for each position in the human genome and for each ENCODE and Roadmap tissue are available online (see Web Resources).
Keywords: functional genomics; noncoding variation; prediction of functional effect.
Copyright © 2018 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Lindblad-Toh K., Garber M., Zuk O., Lin M.F., Parker B.J., Washietl S., Kheradpour P., Ernst J., Jordan G., Mauceli E., Broad Institute Sequencing Platform and Whole Genome Assembly Team. Baylor College of Medicine Human Genome Sequencing Center Sequencing Team. Genome Institute at Washington University A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. - PMC - PubMed
-
- Khurana E., Fu Y., Chakravarty D., Demichelis F., Rubin M.A., Gerstein M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016;17:93–108. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
