

Systematic approach for dissecting the molecular mechanisms of transcriptional regulation in bacteria
- PMID: 29728462
- PMCID: PMC6003448
- DOI: 10.1073/pnas.1722055115
Systematic approach for dissecting the molecular mechanisms of transcriptional regulation in bacteria
Abstract
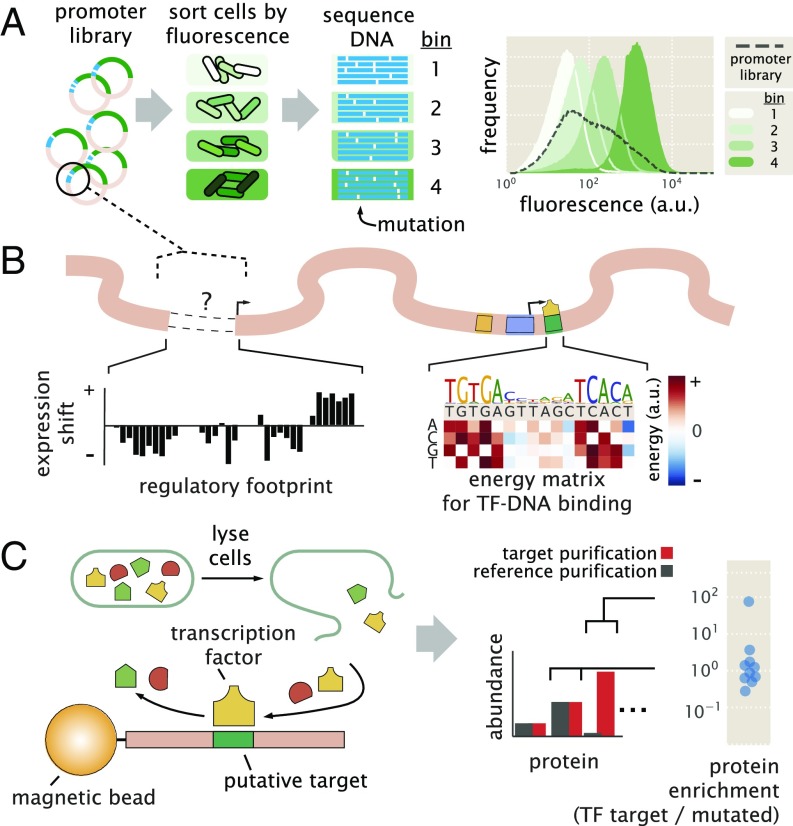
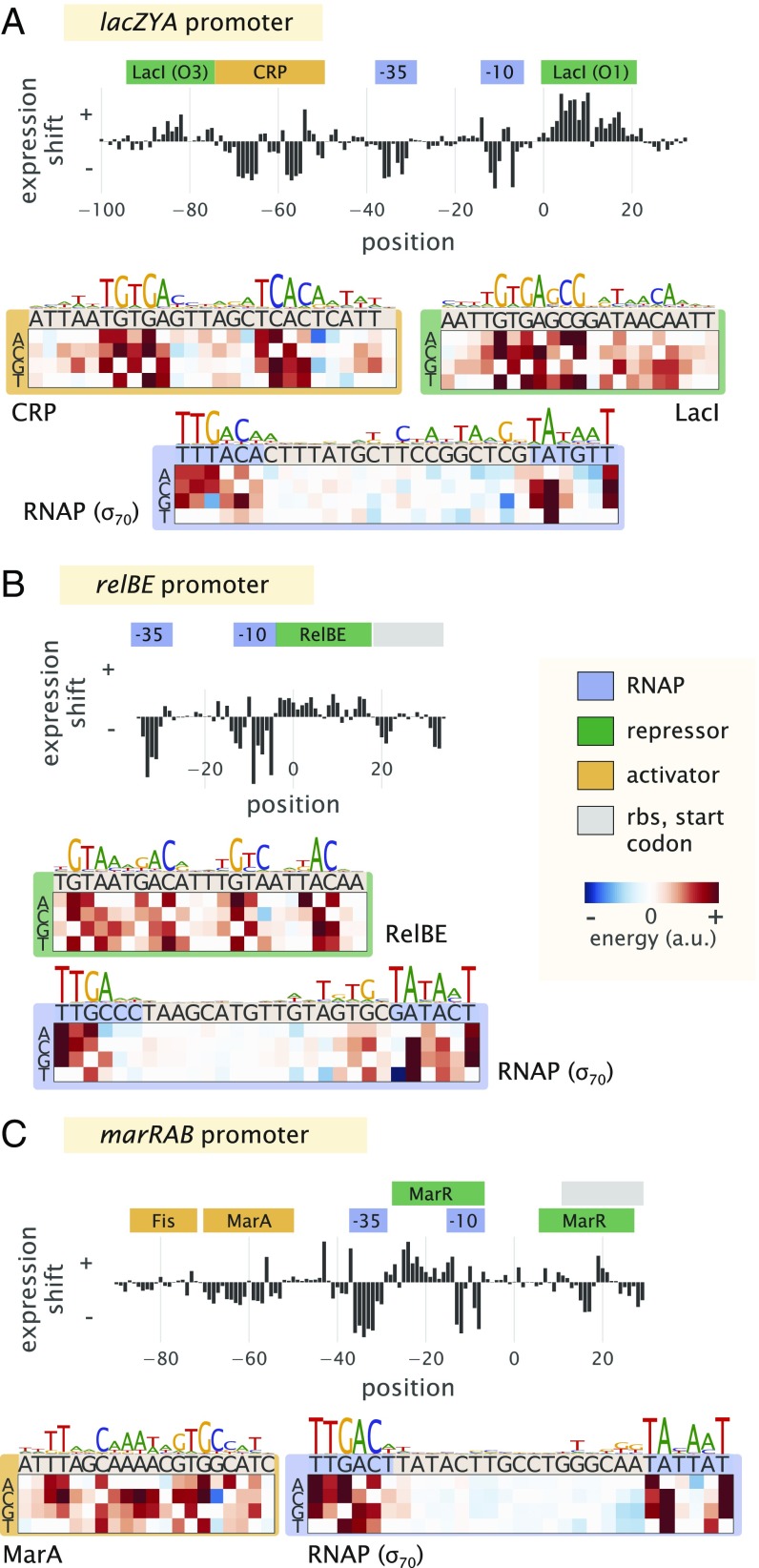
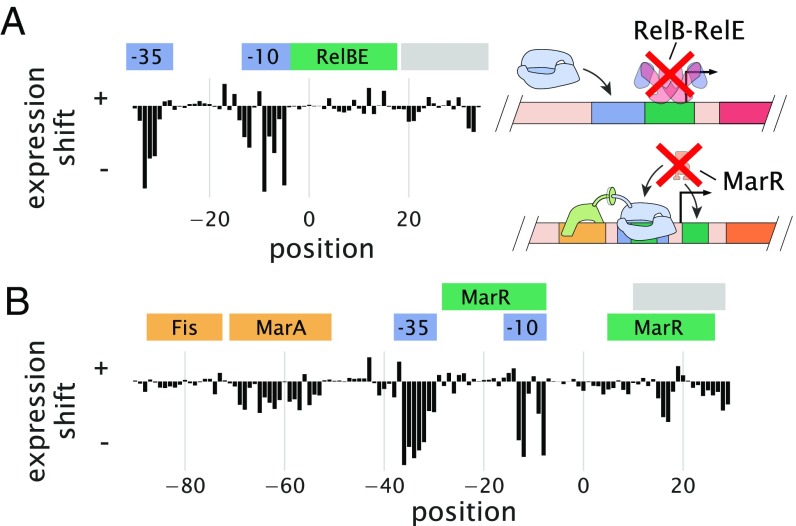
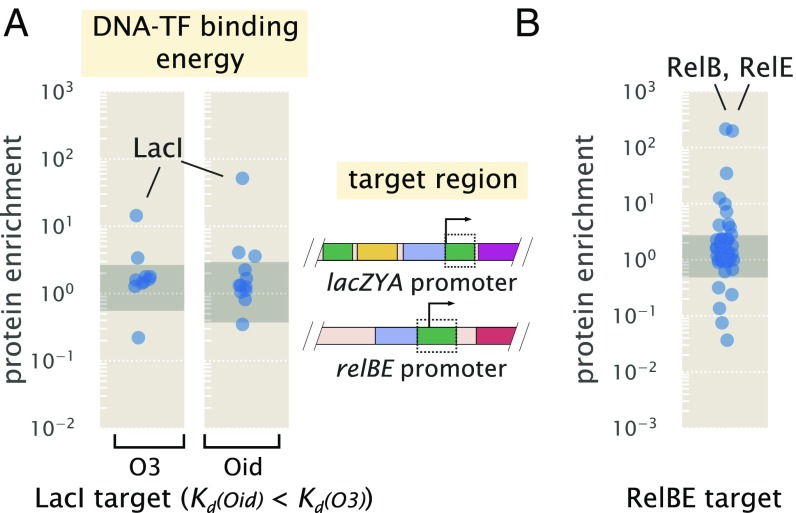
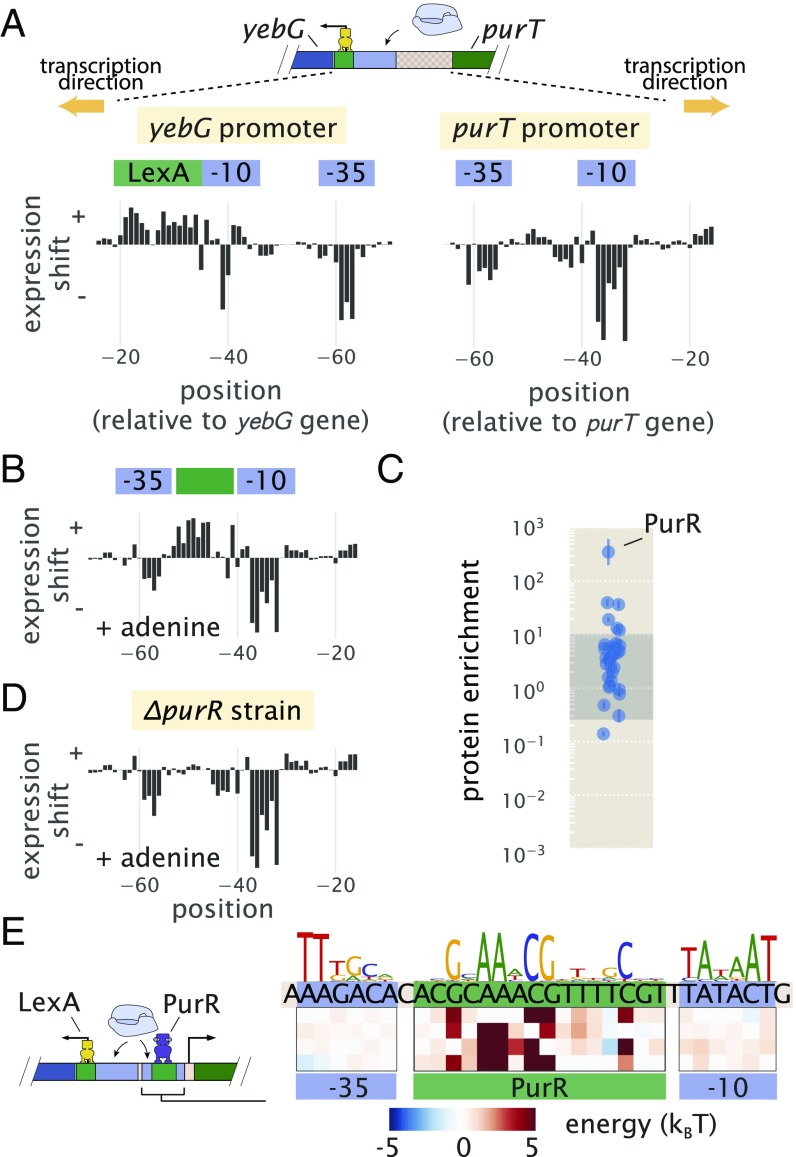
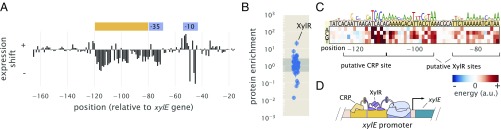
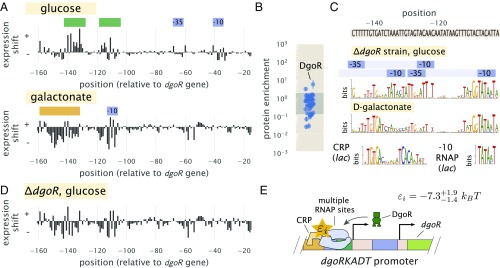
Gene regulation is one of the most ubiquitous processes in biology. However, while the catalog of bacterial genomes continues to expand rapidly, we remain ignorant about how almost all of the genes in these genomes are regulated. At present, characterizing the molecular mechanisms by which individual regulatory sequences operate requires focused efforts using low-throughput methods. Here, we take a first step toward multipromoter dissection and show how a combination of massively parallel reporter assays, mass spectrometry, and information-theoretic modeling can be used to dissect multiple bacterial promoters in a systematic way. We show this approach on both well-studied and previously uncharacterized promoters in the enteric bacterium Escherichia coli In all cases, we recover nucleotide-resolution models of promoter mechanism. For some promoters, including previously unannotated ones, the approach allowed us to further extract quantitative biophysical models describing input-output relationships. Given the generality of the approach presented here, it opens up the possibility of quantitatively dissecting the mechanisms of promoter function in E. coli and a wide range of other bacteria.
Keywords: DNA affinity chromatography; gene regulation; mass spectrometry; massively parallel reporter assay; quantitative models.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
