

Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System
- PMID: 29731704
- PMCID: PMC5920049
- DOI: 10.3389/fnins.2018.00214
Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System
Abstract
Although nerve cell death is the hallmark of many neurological diseases, the processes underlying this death are still poorly defined. However, there is a general consensus that neuronal cell death predominantly proceeds by regulated processes. Almost 30 years ago, a cell death pathway eventually named oxytosis was described in neuronal cells that involved glutathione depletion, reactive oxygen species production, lipoxygenase activation, and calcium influx. More recently, a cell death pathway that involved many of the same steps was described in tumor cells and termed ferroptosis due to a dependence on iron. Since then there has been a great deal of discussion in the literature about whether these are two distinct pathways or cell type- and insult-dependent variations on the same pathway. In this review, we compare and contrast in detail the commonalities and distinctions between the two pathways concluding that the molecular pathways involved in the regulation of ferroptosis and oxytosis are highly similar if not identical. Thus, we suggest that oxytosis and ferroptosis should be regarded as two names for the same cell death pathway. In addition, we describe the potential physiological relevance of oxytosis/ferroptosis in multiple neurological diseases.
Keywords: brain diseases; ferroptosis; iron; oxidative stress; oxytosis; programmed cell death.
Figures
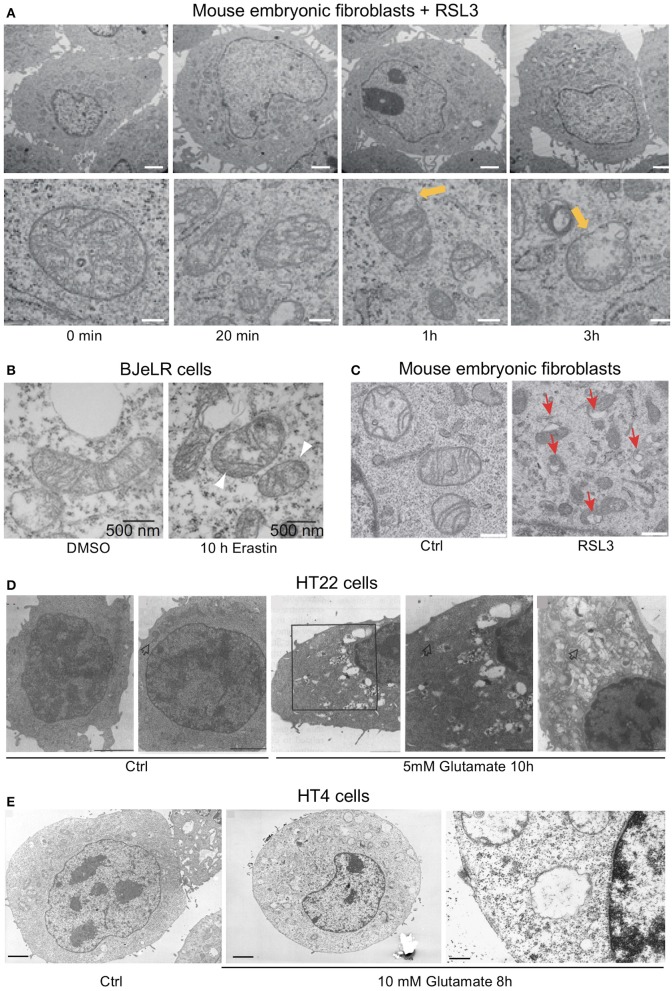
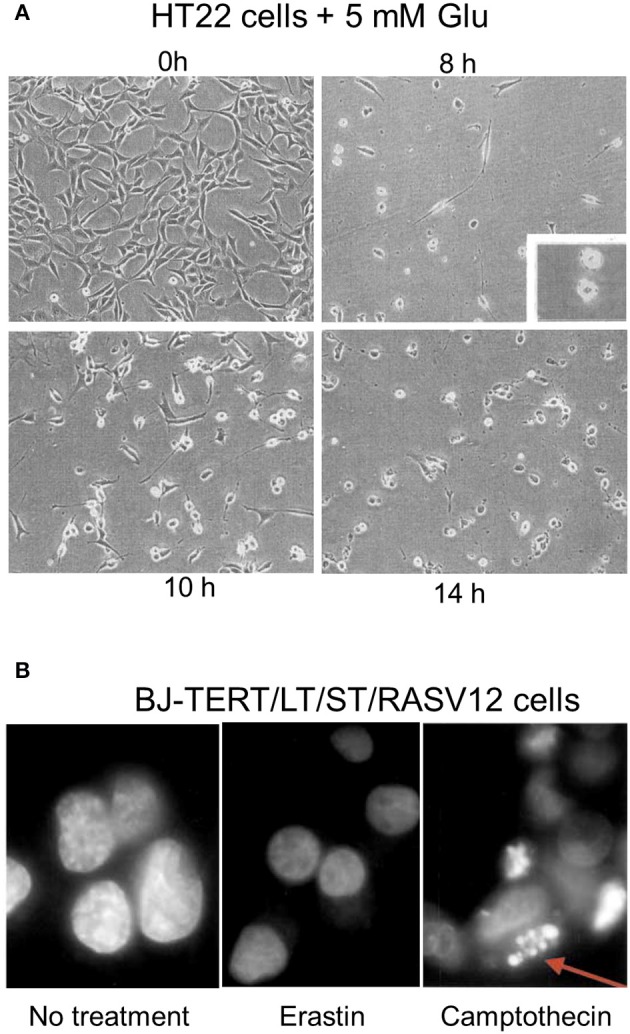
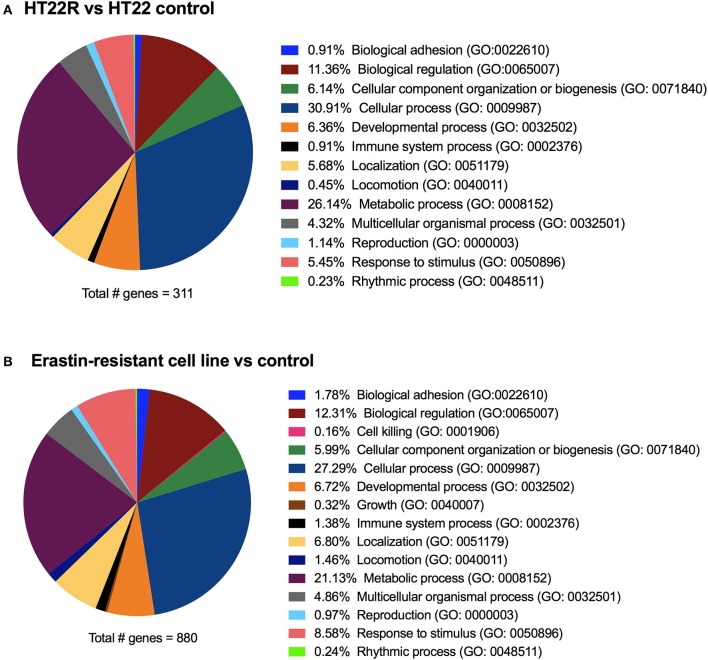
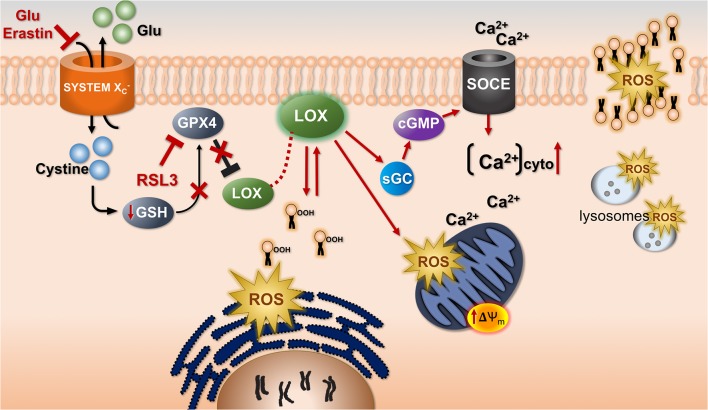
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources