

Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3
- PMID: 29734811
- PMCID: PMC5994137
- DOI: 10.1021/acs.jproteome.8b00217
Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3
Abstract
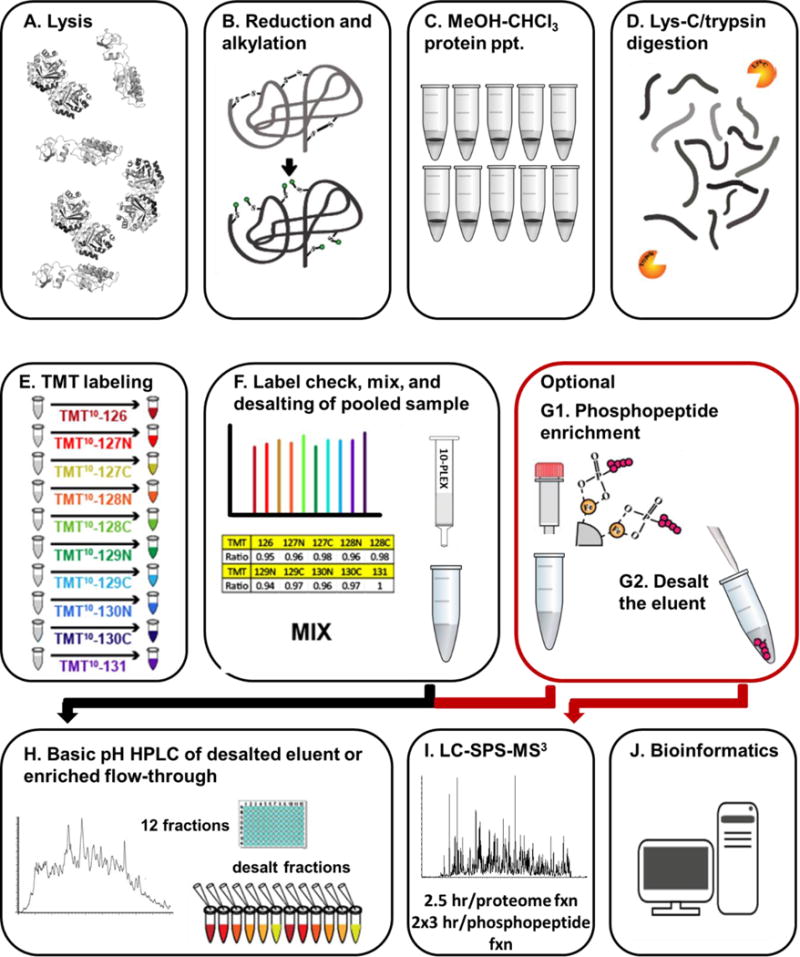
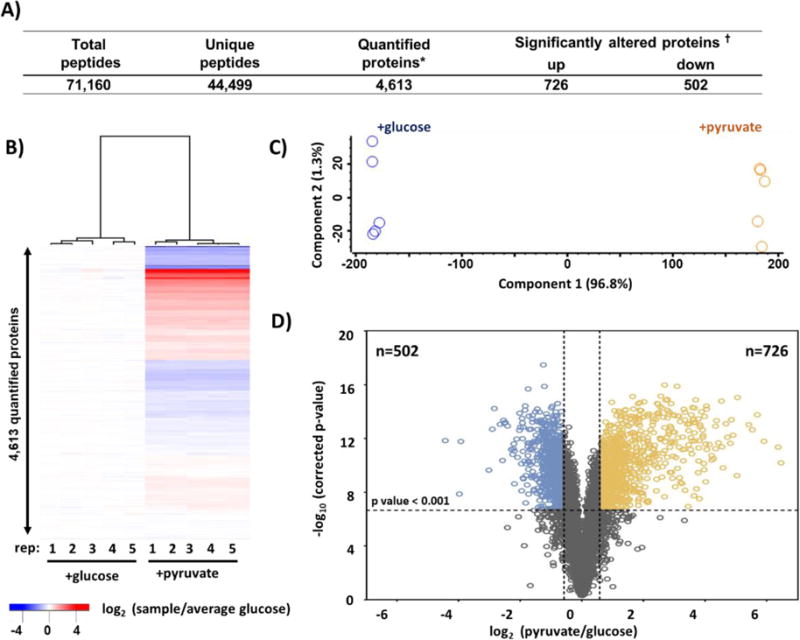
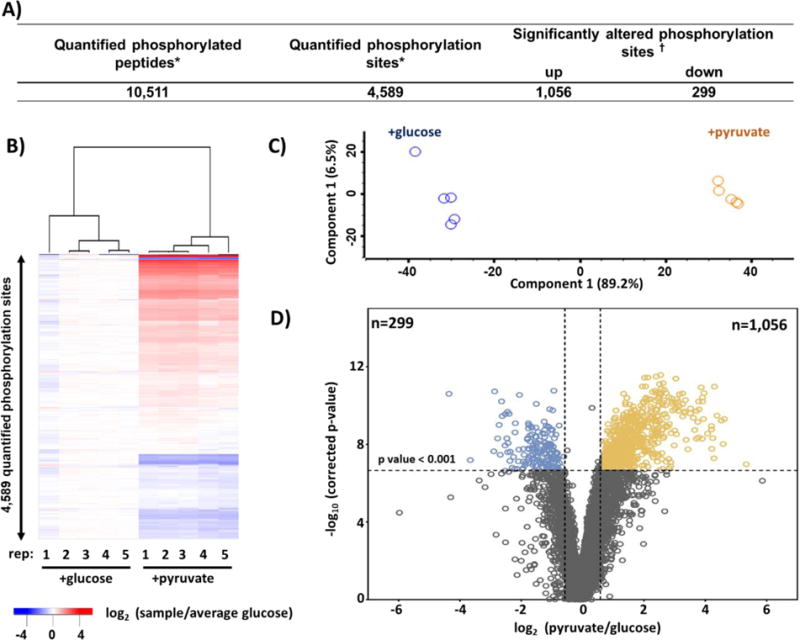
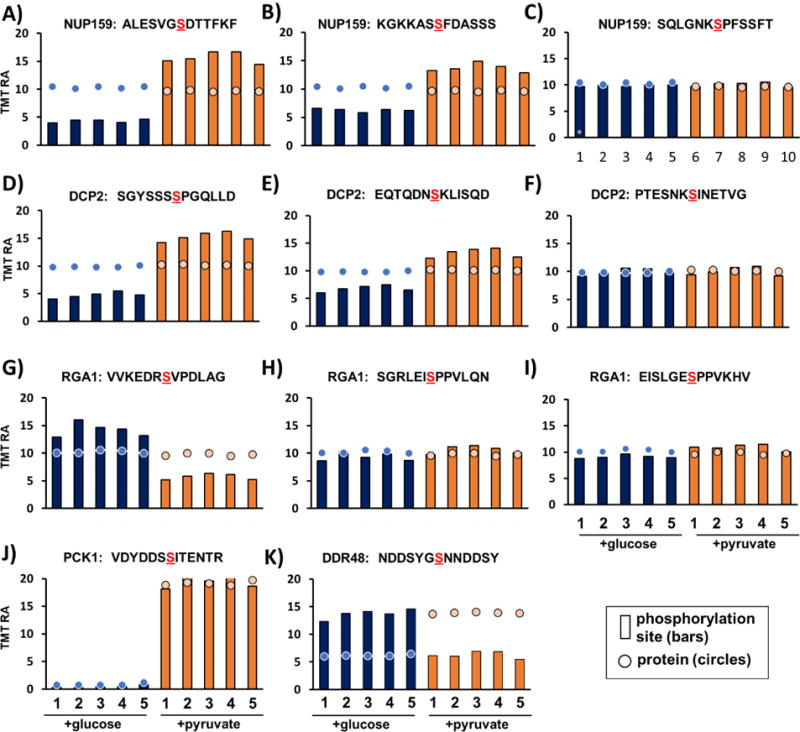
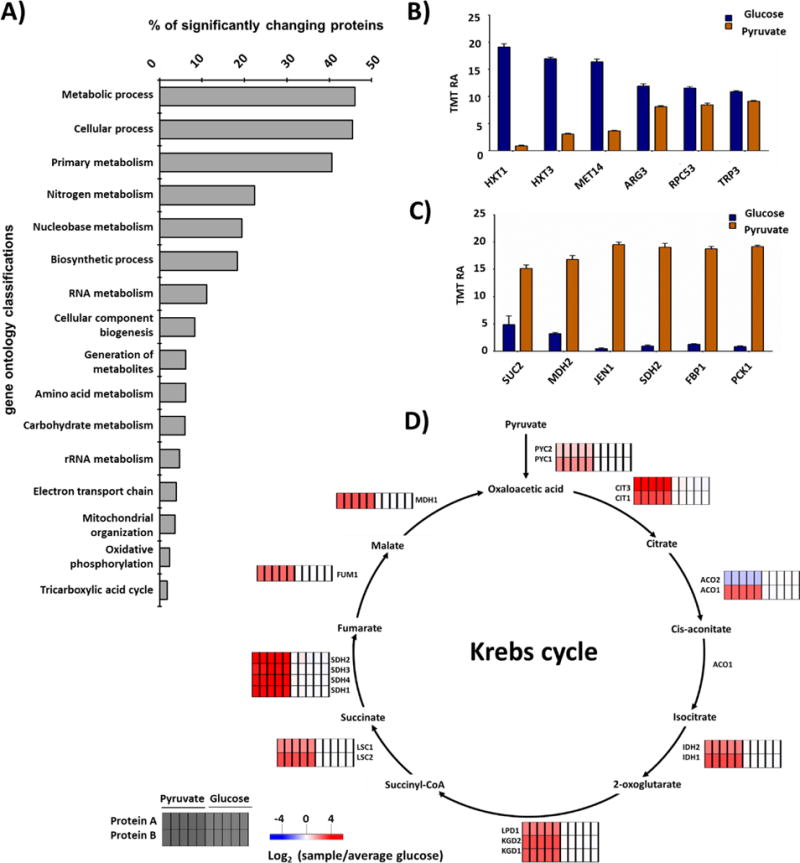
Mass spectrometry (MS) coupled toisobaric labeling has developed rapidly into a powerful strategy for high-throughput protein quantification. Sample multiplexing and exceptional sensitivity allow for the quantification of tens of thousands of peptides and, by inference, thousands of proteins from multiple samples in a single MS experiment. Accurate quantification demands a consistent and robust sample-preparation strategy. Here, we present a detailed workflow for SPS-MS3-based quantitative abundance profiling of tandem mass tag (TMT)-labeled proteins and phosphopeptides that we have named the streamlined (SL)-TMT protocol. We describe a universally applicable strategy that requires minimal individual sample processing and permits the seamless addition of a phosphopeptide enrichment step ("mini-phos") with little deviation from the deep proteome analysis. To showcase our workflow, we profile the proteome of wild-type Saccharomyces cerevisiae yeast grown with either glucose or pyruvate as the carbon source. Here, we have established a streamlined TMT protocol that enables deep proteome and medium-scale phosphoproteome analysis.
Keywords: Orbitrap Fusion Lumos; SPS; multi-notch; phosphoproteome; sample preparation; synchronous precursor selection; tandem mass tag.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. - PubMed
-
- Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75(8):1895–904. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
