

Machine learning identifies signatures of host adaptation in the bacterial pathogen Salmonella enterica
- PMID: 29738521
- PMCID: PMC5940178
- DOI: 10.1371/journal.pgen.1007333
Machine learning identifies signatures of host adaptation in the bacterial pathogen Salmonella enterica
Abstract
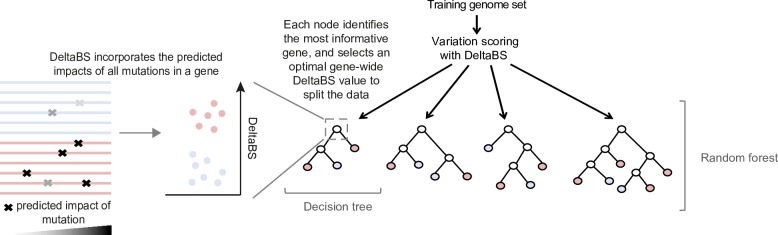
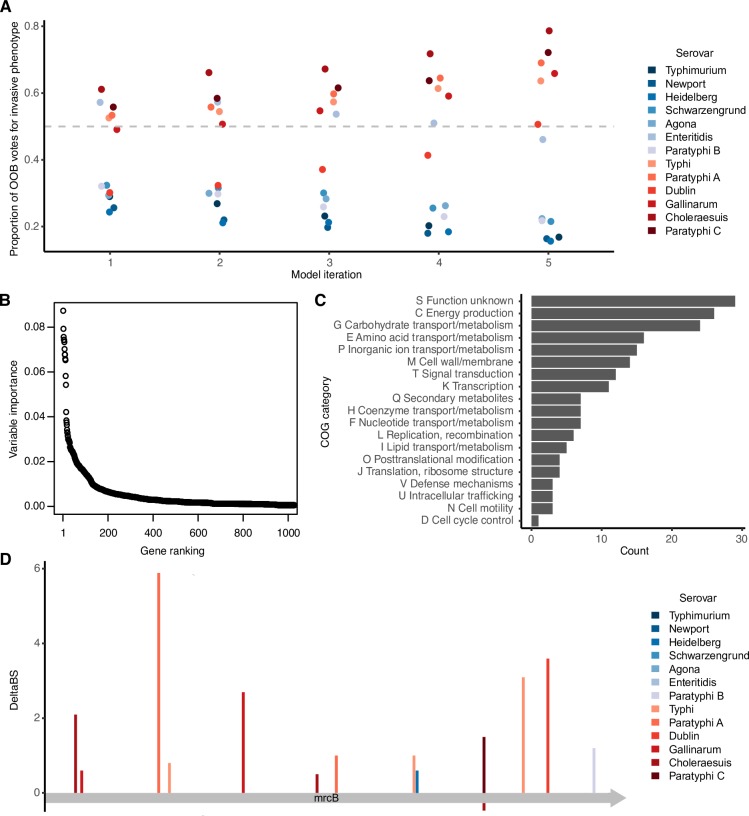
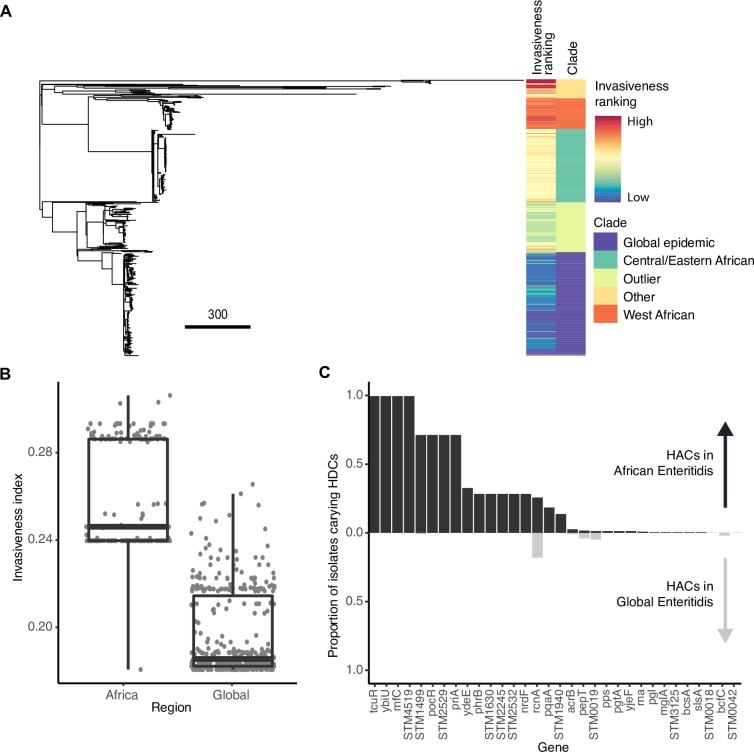
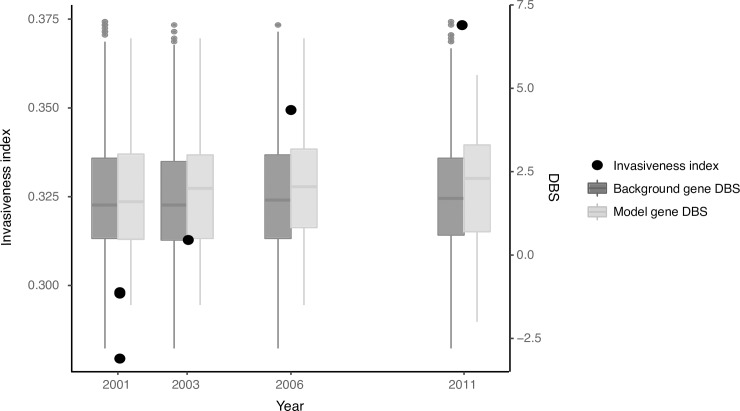
Emerging pathogens are a major threat to public health, however understanding how pathogens adapt to new niches remains a challenge. New methods are urgently required to provide functional insights into pathogens from the massive genomic data sets now being generated from routine pathogen surveillance for epidemiological purposes. Here, we measure the burden of atypical mutations in protein coding genes across independently evolved Salmonella enterica lineages, and use these as input to train a random forest classifier to identify strains associated with extraintestinal disease. Members of the species fall along a continuum, from pathovars which cause gastrointestinal infection and low mortality, associated with a broad host-range, to those that cause invasive infection and high mortality, associated with a narrowed host range. Our random forest classifier learned to perfectly discriminate long-established gastrointestinal and invasive serovars of Salmonella. Additionally, it was able to discriminate recently emerged Salmonella Enteritidis and Typhimurium lineages associated with invasive disease in immunocompromised populations in sub-Saharan Africa, and within-host adaptation to invasive infection. We dissect the architecture of the model to identify the genes that were most informative of phenotype, revealing a common theme of degradation of metabolic pathways in extraintestinal lineages. This approach accurately identifies patterns of gene degradation and diversifying selection specific to invasive serovars that have been captured by more labour-intensive investigations, but can be readily scaled to larger analyses.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Frank SA, Schmid-Hempel P. Mechanisms of pathogenesis and the evolution of parasite virulence. J Evol Biol. 2008;21: 396–404. doi: 10.1111/j.1420-9101.2007.01480.x - DOI - PubMed
-
- Fauci AS, Morens DM. The perpetual challenge of infectious diseases. N Engl J Med. 2012;366: 454–461. doi: 10.1056/NEJMra1108296 - DOI - PubMed
-
- Pallen MJ, Wren BW. Bacterial pathogenomics. Nature. nature.com; 2007;449: 835–842. doi: 10.1038/nature06248 - DOI - PubMed
-
- Loman NJ, Pallen MJ. Twenty years of bacterial genome sequencing. Nat Rev Microbiol. 2015;13: 787–794. doi: 10.1038/nrmicro3565 - DOI - PubMed
-
- McNally A, Thomson NR, Reuter S, Wren BW. “Add, stir and reduce”: Yersinia spp. as model bacteria for pathogen evolution. Nat Rev Microbiol. 2016;14: 177–190. doi: 10.1038/nrmicro.2015.29 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
