

MAPK Reliance via Acquired CDK4/6 Inhibitor Resistance in Cancer
- PMID: 29739788
- PMCID: PMC6125187
- DOI: 10.1158/1078-0432.CCR-18-0410
MAPK Reliance via Acquired CDK4/6 Inhibitor Resistance in Cancer
Abstract
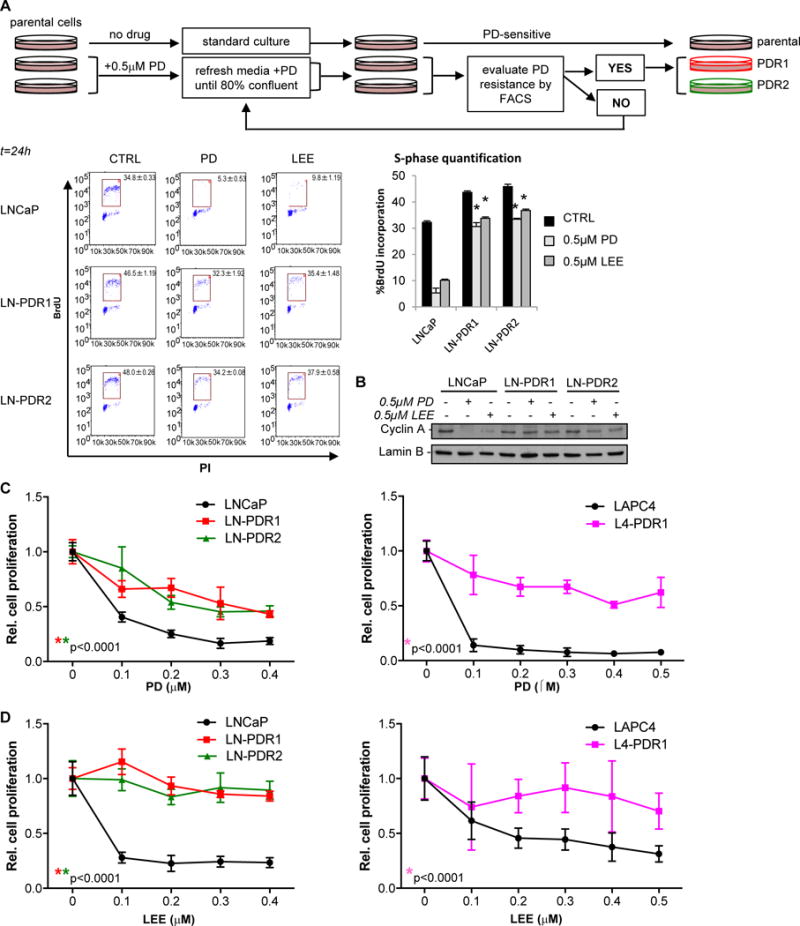
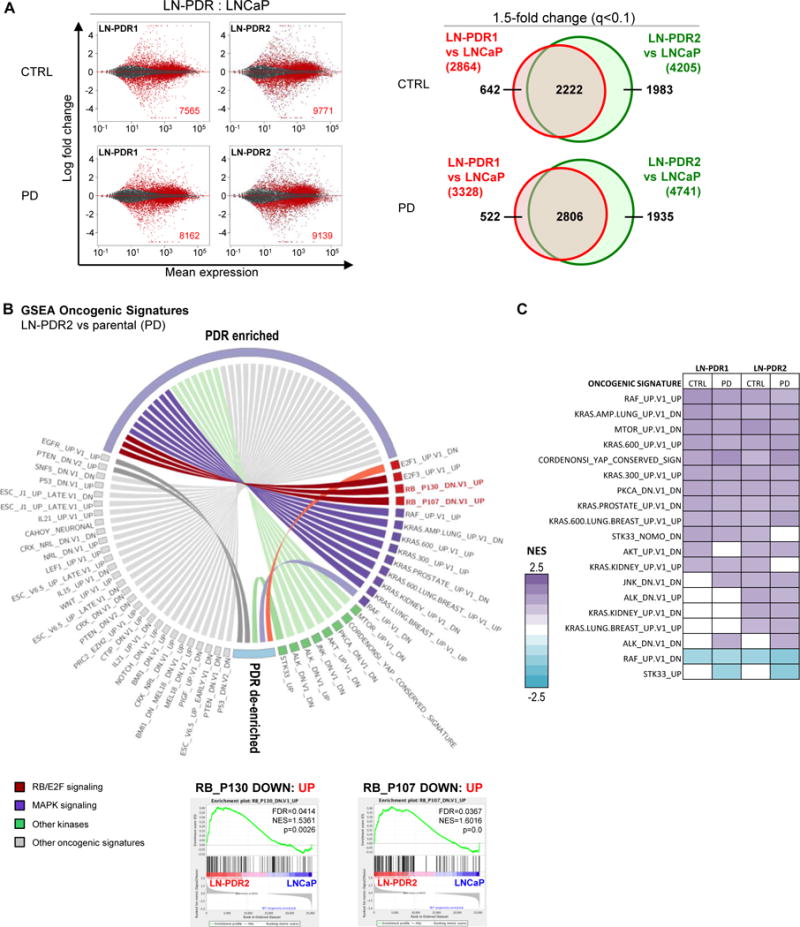
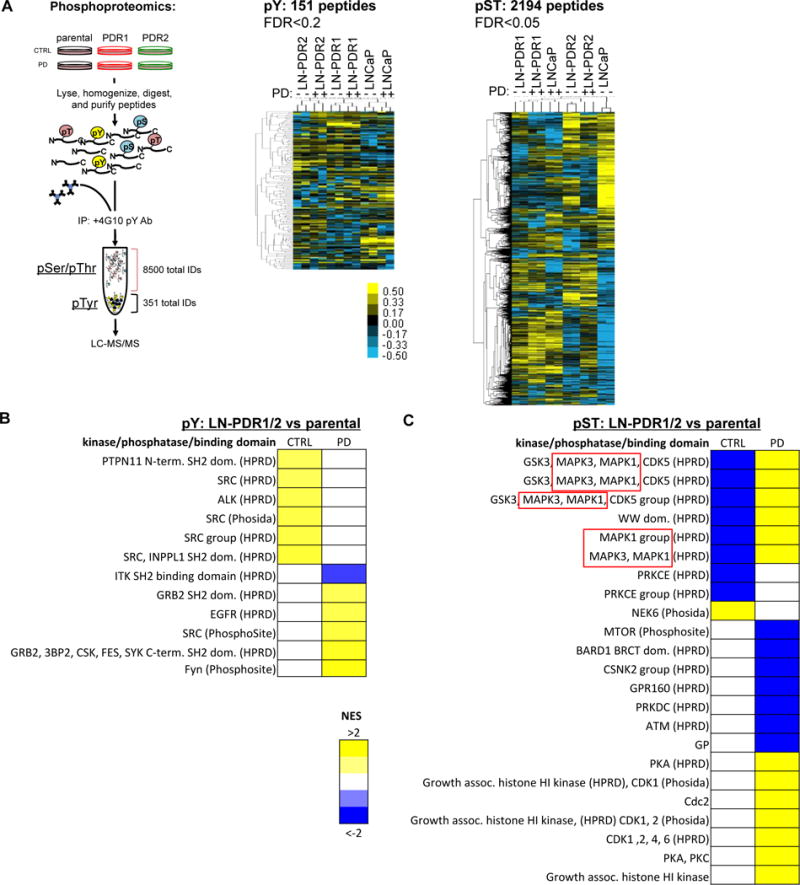
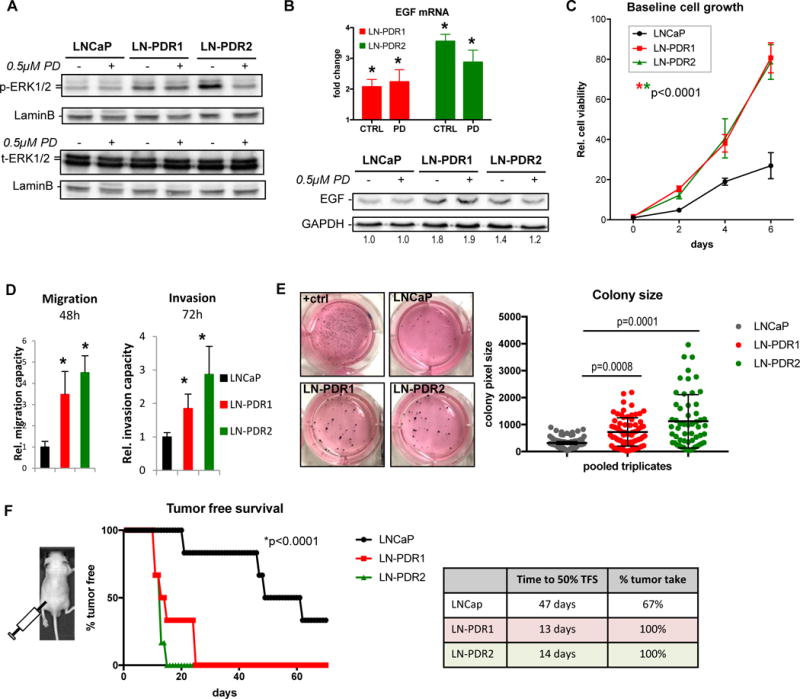
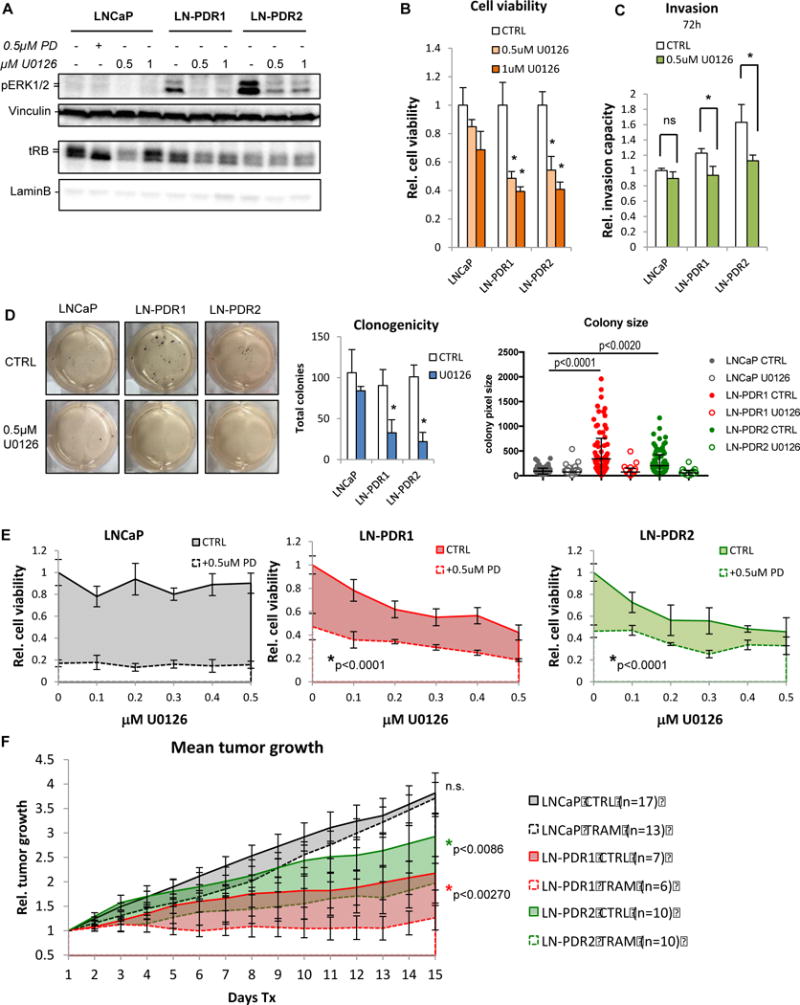
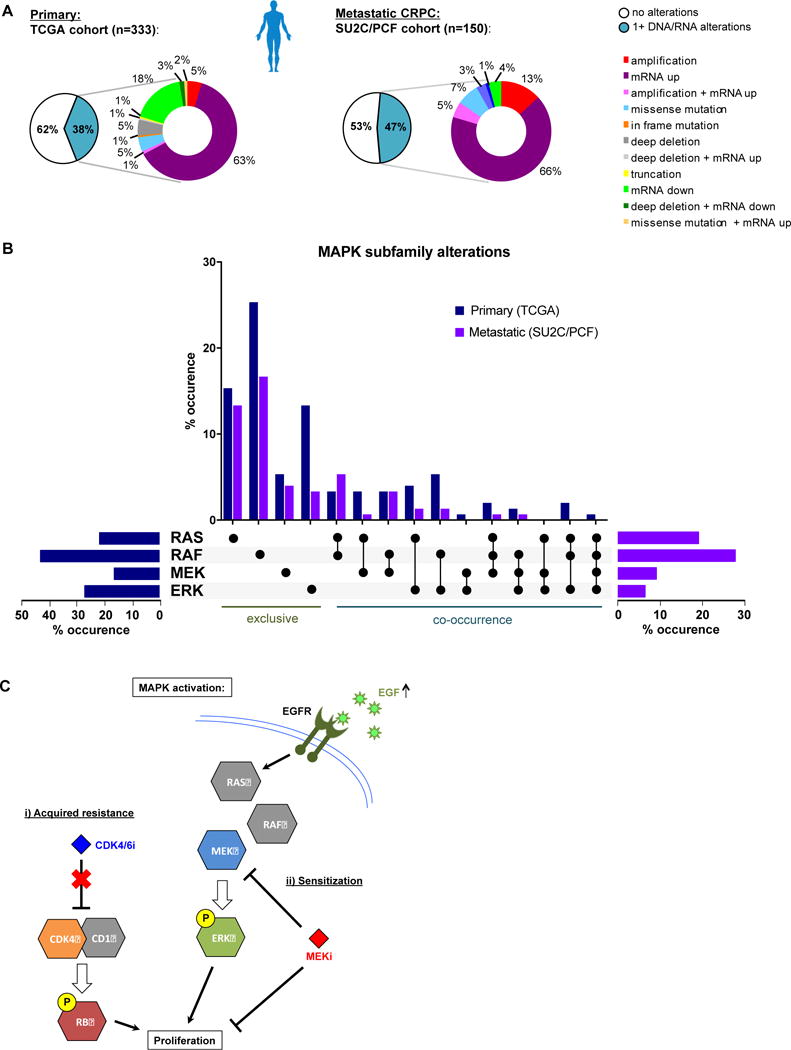
Purpose: Loss of cell-cycle control is a hallmark of cancer, which can be targeted with agents, including cyclin-dependent kinase-4/6 (CDK4/6) kinase inhibitors that impinge upon the G1-S cell-cycle checkpoint via maintaining activity of the retinoblastoma tumor suppressor (RB). This class of drugs is under clinical investigation for various solid tumor types and has recently been FDA-approved for treatment of breast cancer. However, development of therapeutic resistance is not uncommon.Experimental Design: In this study, palbociclib (a CDK4/6 inhibitor) resistance was established in models of early stage, RB-positive cancer.Results: This study demonstrates that acquired palbociclib resistance renders cancer cells broadly resistant to CDK4/6 inhibitors. Acquired resistance was associated with aggressive in vitro and in vivo phenotypes, including proliferation, migration, and invasion. Integration of RNA sequencing analysis and phosphoproteomics profiling revealed rewiring of the kinome, with a strong enrichment for enhanced MAPK signaling across all resistance models, which resulted in aggressive in vitro and in vivo phenotypes and prometastatic signaling. However, CDK4/6 inhibitor-resistant models were sensitized to MEK inhibitors, revealing reliance on active MAPK signaling to promote tumor cell growth and invasion.Conclusions: In sum, these studies identify MAPK reliance in acquired CDK4/6 inhibitor resistance that promotes aggressive disease, while nominating MEK inhibition as putative novel therapeutic strategy to treat or prevent CDK4/6 inhibitor resistance in cancer. Clin Cancer Res; 24(17); 4201-14. ©2018 AACR.
©2018 American Association for Cancer Research.
Conflict of interest statement
Figures
References
-
- Vijayaraghavan S, Moulder S, Keyomarsi K, Layman RM. Inhibiting CDK in Cancer Therapy: Current Evidence and Future Directions. Target Oncol. 2017 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
