

Interleukin-1 β Activates a MYC-Dependent Metabolic Switch in Kidney Stromal Cells Necessary for Progressive Tubulointerstitial Fibrosis
- PMID: 29739813
- PMCID: PMC6054344
- DOI: 10.1681/ASN.2017121283
Interleukin-1 β Activates a MYC-Dependent Metabolic Switch in Kidney Stromal Cells Necessary for Progressive Tubulointerstitial Fibrosis
Abstract
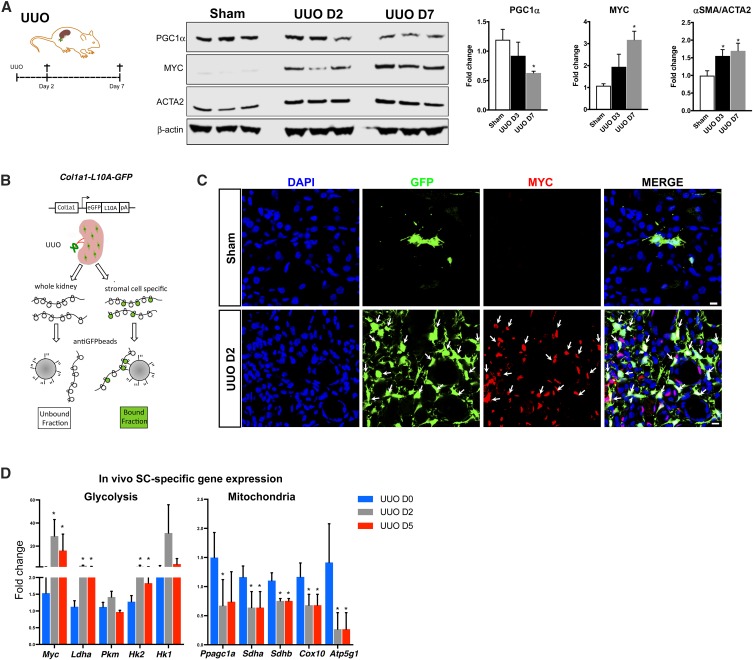
Background Kidney injury is characterized by persisting inflammation and fibrosis, yet mechanisms by which inflammatory signals drive fibrogenesis remain poorly defined.Methods RNA sequencing of fibrotic kidneys from patients with CKD identified a metabolic gene signature comprising loss of mitochondrial and oxidative phosphorylation gene expression with a concomitant increase in regulators and enzymes of glycolysis under the control of PGC1α and MYC transcription factors, respectively. We modeled this metabolic switch in vivo, in experimental murine models of kidney injury, and in vitro in human kidney stromal cells (SCs) and human kidney organoids.Results In mice, MYC and the target genes thereof became activated in resident SCs early after kidney injury, suggesting that acute innate immune signals regulate this transcriptional switch. In vitro, stimulation of purified human kidney SCs and human kidney organoids with IL-1β recapitulated the molecular events observed in vivo, inducing functional metabolic derangement characterized by increased MYC-dependent glycolysis, the latter proving necessary to drive proliferation and matrix production. MYC interacted directly with sequestosome 1/p62, which is involved in proteasomal degradation, and modulation of p62 expression caused inverse effects on MYC expression. IL-1β stimulated autophagy flux, causing degradation of p62 and accumulation of MYC. Inhibition of the IL-1R signal transducer kinase IRAK4 in vivo or inhibition of MYC in vivo as well as in human kidney organoids in vitro abrogated fibrosis and reduced tubular injury.Conclusions Our findings define a connection between IL-1β and metabolic switch in fibrosis initiation and progression and highlight IL-1β and MYC as potential therapeutic targets in tubulointerstitial diseases.
Keywords: Cell Signaling; Chronic inflammation; interstitial fibrosis.
Copyright © 2018 by the American Society of Nephrology.
Figures







Comment in
-
IL-1β switches on kidney fibrosis.Nat Rev Nephrol. 2018 Aug;14(8):475. doi: 10.1038/s41581-018-0026-2. Nat Rev Nephrol. 2018. PMID: 29799011 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
