

Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics
- PMID: 29739930
- PMCID: PMC5940825
- DOI: 10.1038/s41467-018-03621-1
Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics
Abstract
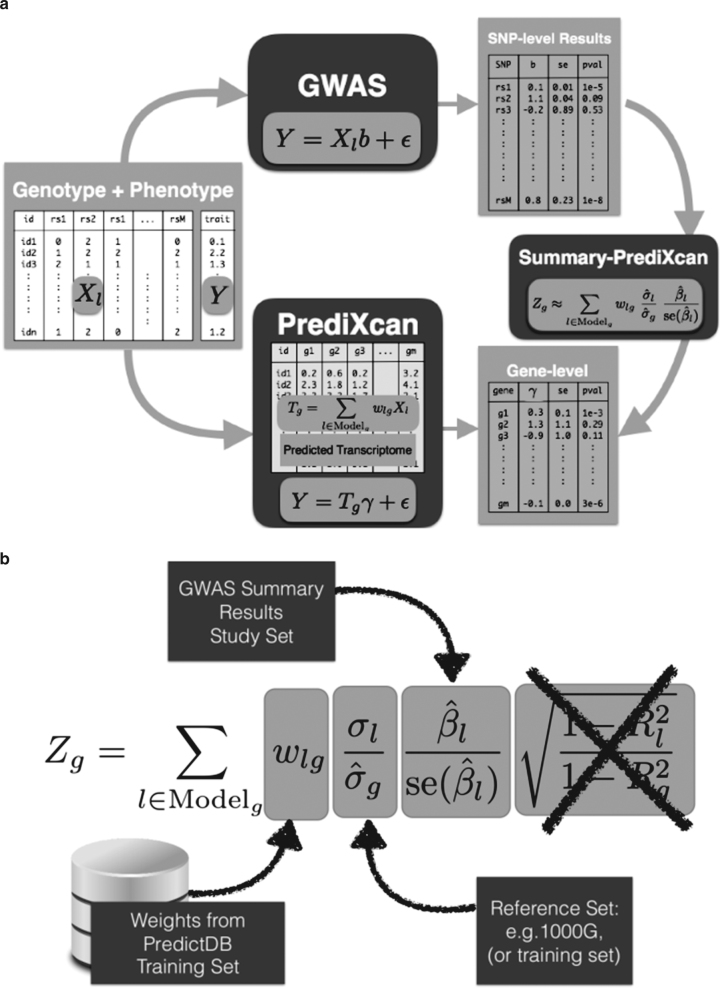
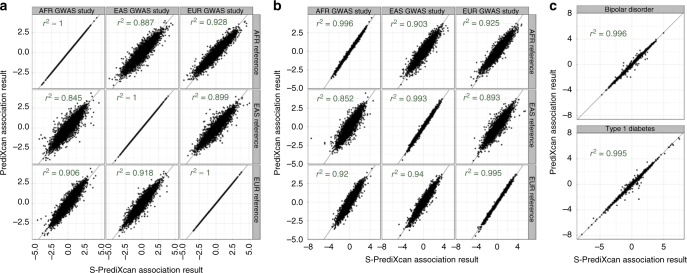
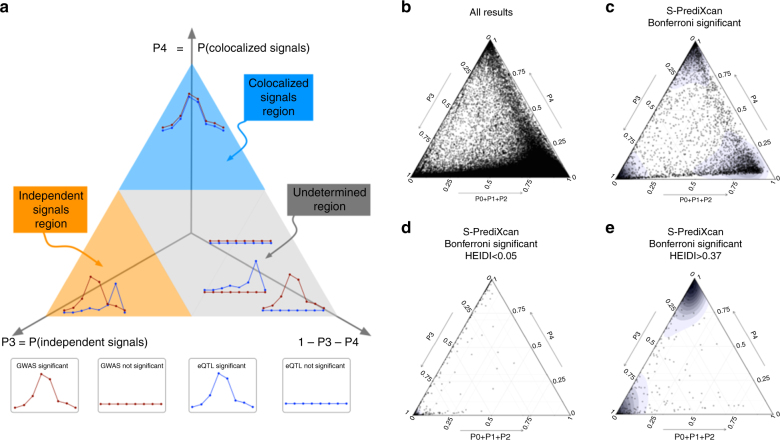
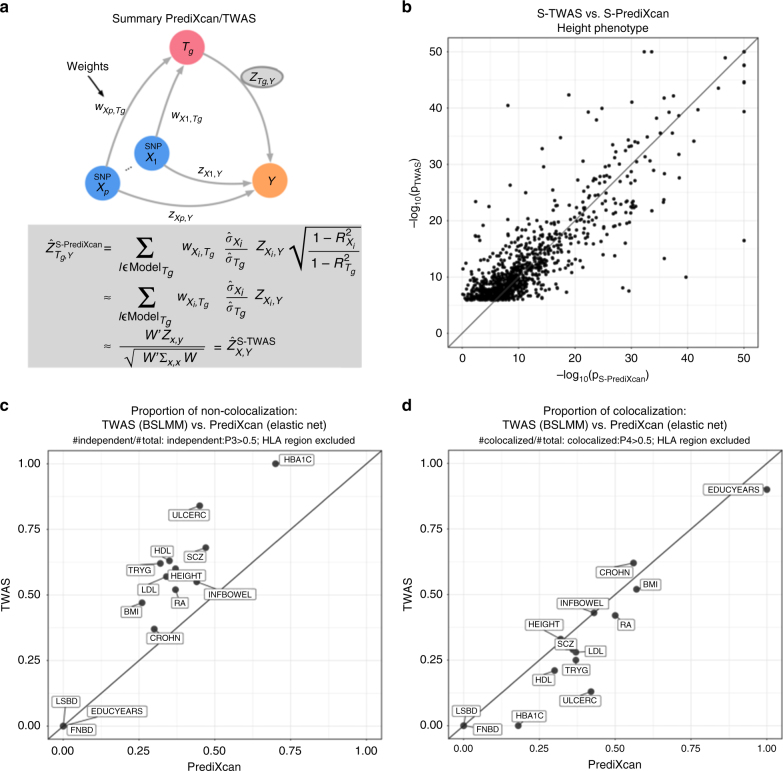
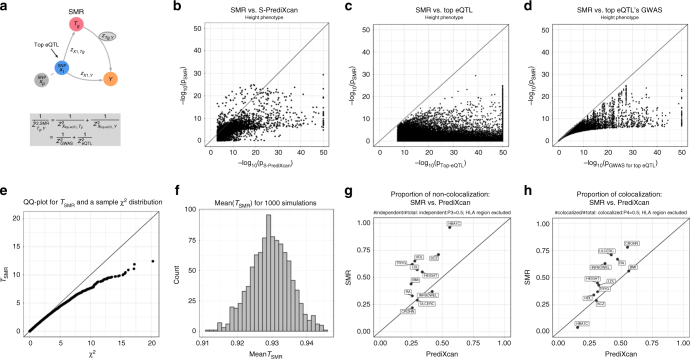
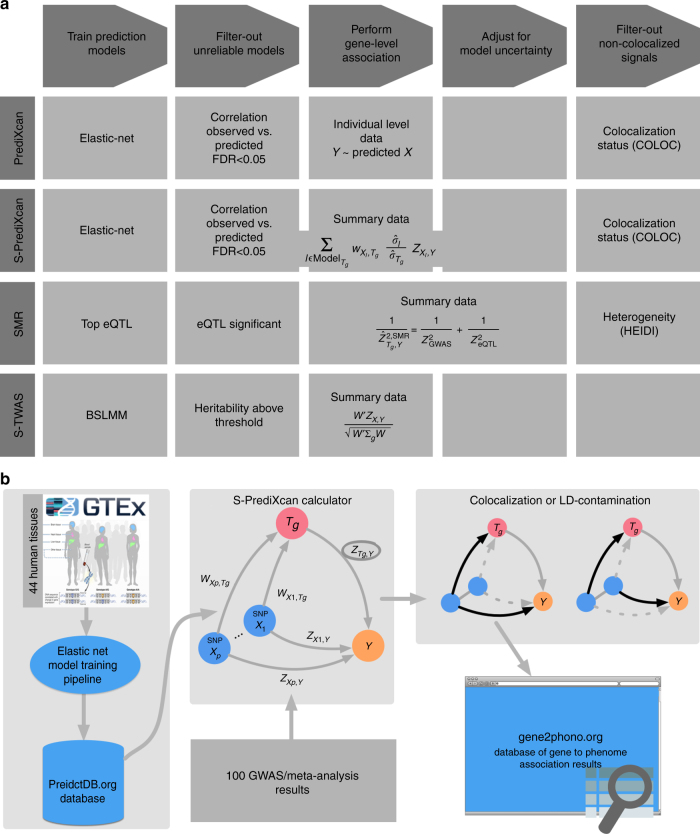
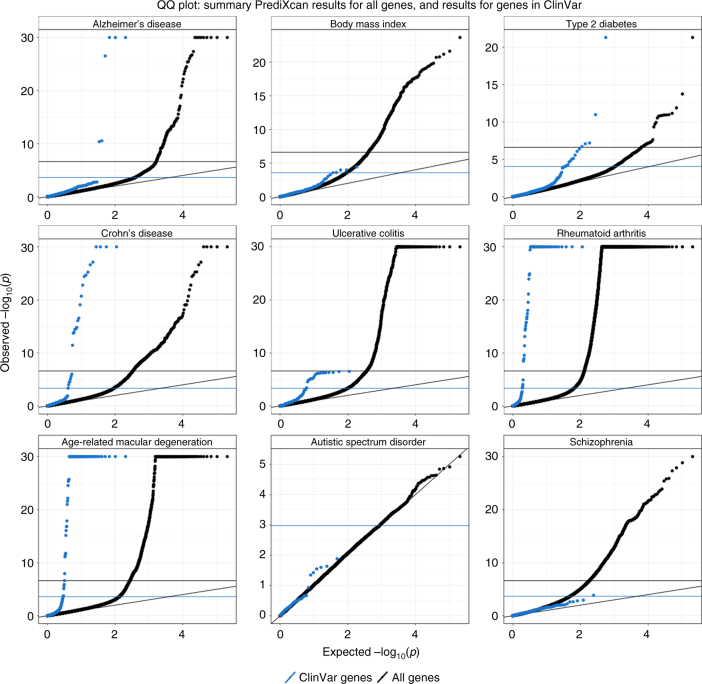
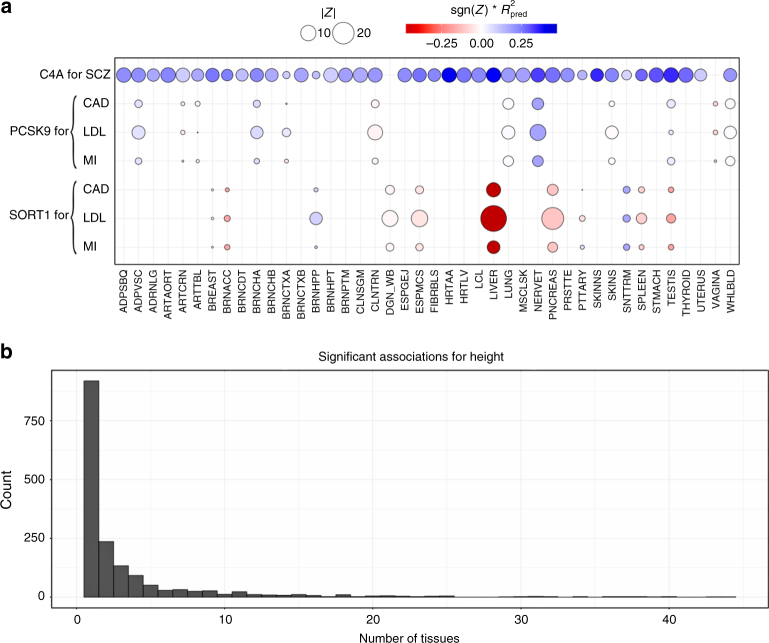
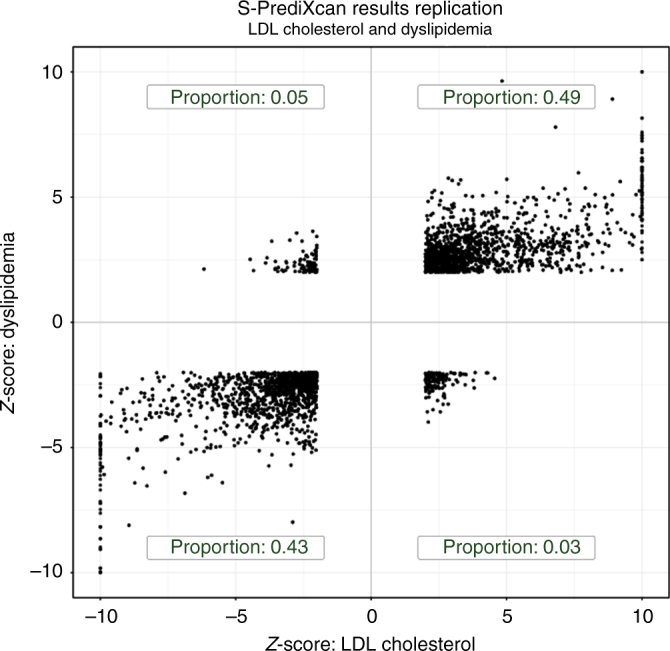
Scalable, integrative methods to understand mechanisms that link genetic variants with phenotypes are needed. Here we derive a mathematical expression to compute PrediXcan (a gene mapping approach) results using summary data (S-PrediXcan) and show its accuracy and general robustness to misspecified reference sets. We apply this framework to 44 GTEx tissues and 100+ phenotypes from GWAS and meta-analysis studies, creating a growing public catalog of associations that seeks to capture the effects of gene expression variation on human phenotypes. Replication in an independent cohort is shown. Most of the associations are tissue specific, suggesting context specificity of the trait etiology. Colocalized significant associations in unexpected tissues underscore the need for an agnostic scanning of multiple contexts to improve our ability to detect causal regulatory mechanisms. Monogenic disease genes are enriched among significant associations for related traits, suggesting that smaller alterations of these genes may cause a spectrum of milder phenotypes.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
- R01MH107666 /NH/NIH HHS/United States
- F31 DK101202/DK/NIDDK NIH HHS/United States
- R01 DA006227/DA/NIDA NIH HHS/United States
- R01 MH107666/MH/NIMH NIH HHS/United States
- R01 MH101782/MH/NIMH NIH HHS/United States
- R01 MH101819/MH/NIMH NIH HHS/United States
- T32 MH020065/MH/NIMH NIH HHS/United States
- R01 MH090951/MH/NIMH NIH HHS/United States
- R01 MH101820/MH/NIMH NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 MH101822/MH/NIMH NIH HHS/United States
- R01 MH090937/MH/NIMH NIH HHS/United States
- UL1 TR000430/TR/NCATS NIH HHS/United States
- P50 MH094267 /NH/NIH HHS/United States
- R01 MH101810/MH/NIMH NIH HHS/United States
- P50 MH094267/MH/NIMH NIH HHS/United States
- R01 DA033684/DA/NIDA NIH HHS/United States
- T32 MH020065 /NH/NIH HHS/United States
- P30 DK20595 /NH/NIH HHS/United States
- R01 MH090936/MH/NIMH NIH HHS/United States
- P50 DA037844 /NH/NIH HHS/United States
- R15 HG009569/HG/NHGRI NIH HHS/United States
- P50 DA037844/DA/NIDA NIH HHS/United States
- R01 MH101825/MH/NIMH NIH HHS/United States
- R01 MH090948/MH/NIMH NIH HHS/United States
- R01 MH090941/MH/NIMH NIH HHS/United States
- F31 DK101202 /NH/NIH HHS/United States
- R01 MH101814/MH/NIMH NIH HHS/United States
- P30 DK020595/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
