

Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia
- PMID: 29740158
- PMCID: PMC5990522
- DOI: 10.1038/s41375-018-0127-8
Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia
Abstract
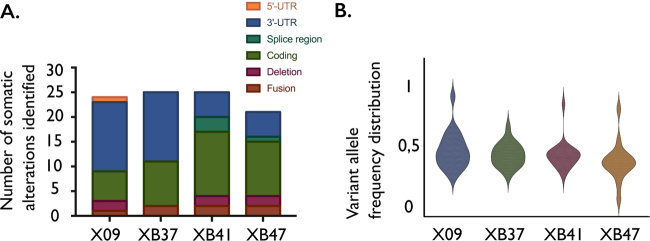
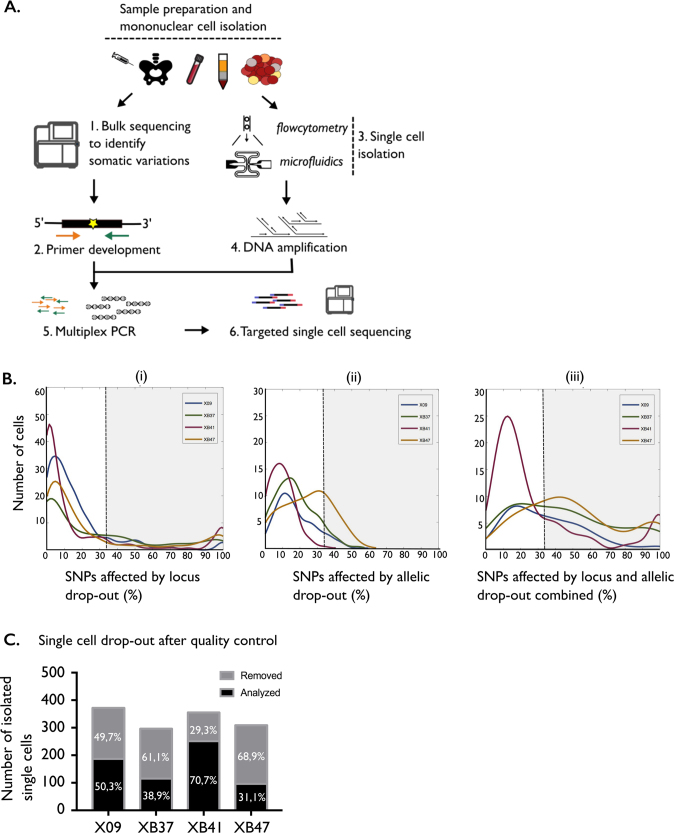
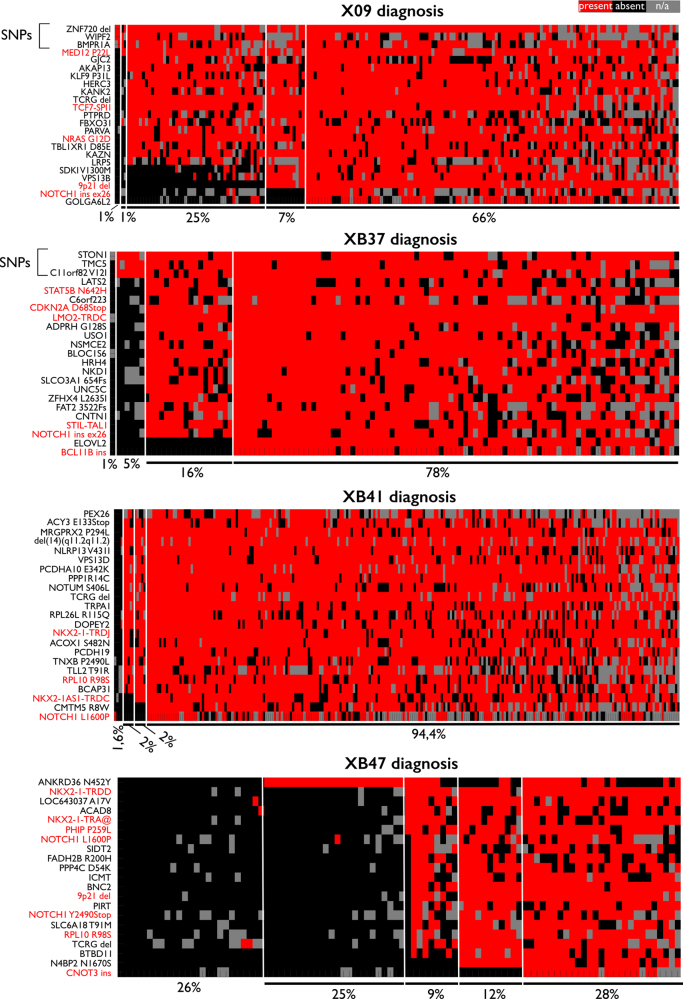
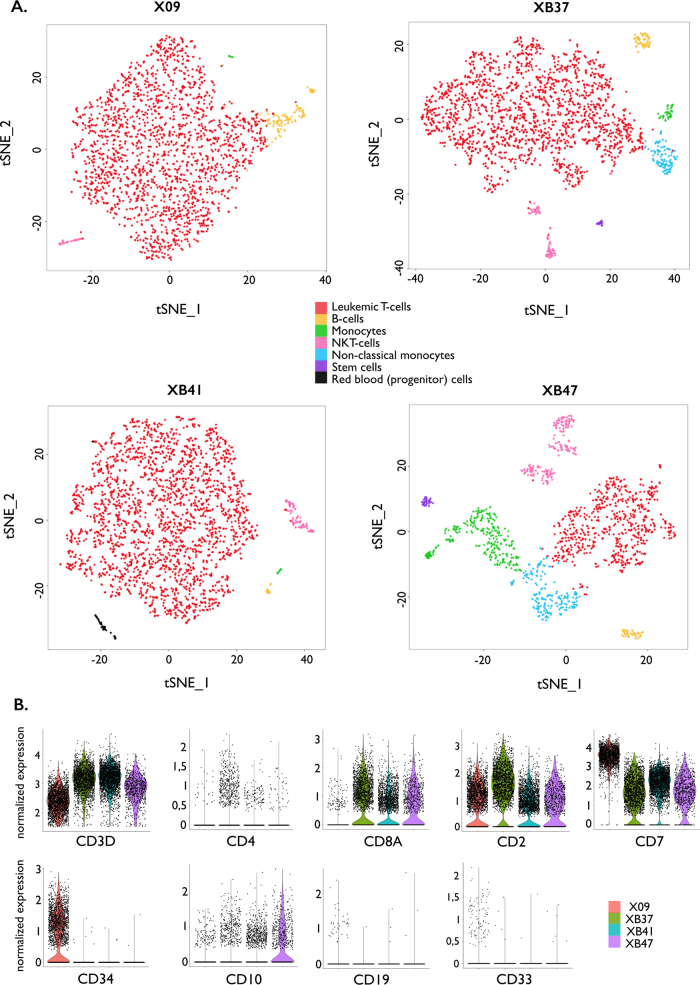
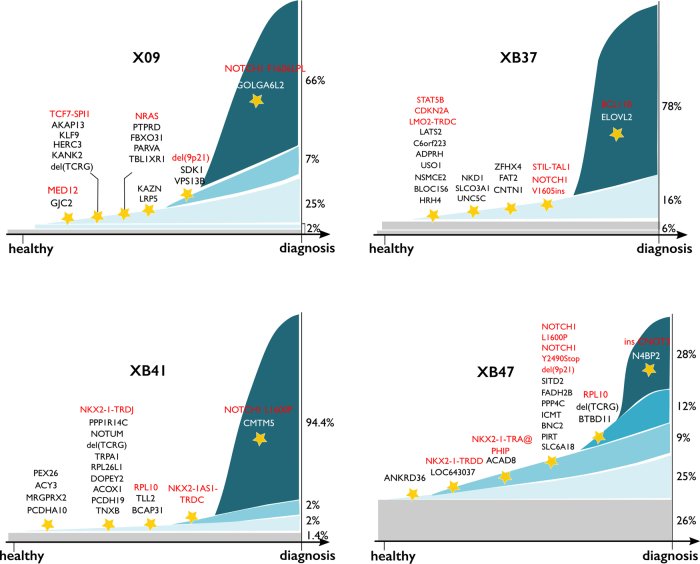
Next-generation sequencing has provided a detailed overview of the various genomic lesions implicated in the pathogenesis of T-cell acute lymphoblastic leukemia (T-ALL). Typically, 10-20 protein-altering lesions are found in T-ALL cells at diagnosis. However, it is currently unclear in which order these mutations are acquired and in which progenitor cells this is initiated. To address these questions, we used targeted single-cell sequencing of total bone marrow cells and CD34+CD38- multipotent progenitor cells for four T-ALL cases. Hierarchical clustering detected a dominant leukemia cluster at diagnosis, accompanied by a few smaller clusters harboring only a fraction of the mutations. We developed a graph-based algorithm to determine the order of mutation acquisition. Two of the four patients had an early event in a known oncogene (MED12, STAT5B) among various pre-leukemic events. Intermediate events included loss of 9p21 (CDKN2A/B) and acquisition of fusion genes, while NOTCH1 mutations were typically late events. Analysis of CD34+CD38- cells and myeloid progenitors revealed that in half of the cases somatic mutations were detectable in multipotent progenitor cells. We demonstrate that targeted single-cell sequencing can elucidate the order of mutation acquisition in T-ALL and that T-ALL development can start in a multipotent progenitor cell.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
