

Genomic DNA Methylation-Derived Algorithm Enables Accurate Detection of Malignant Prostate Tissues
- PMID: 29740534
- PMCID: PMC5925605
- DOI: 10.3389/fonc.2018.00100
Genomic DNA Methylation-Derived Algorithm Enables Accurate Detection of Malignant Prostate Tissues
Abstract
Introduction: The current methodology involving diagnosis of prostate cancer (PCa) relies on the pathology examination of prostate needle biopsies, a method with high false negative rates partly due to temporospatial, molecular, and morphological heterogeneity of prostate adenocarcinoma. It is postulated that molecular markers have a potential to assign diagnosis to a considerable portion of undetected prostate tumors. This study examines the genome-wide DNA methylation changes in PCa in search of genomic markers for the development of a diagnostic algorithm for PCa screening.
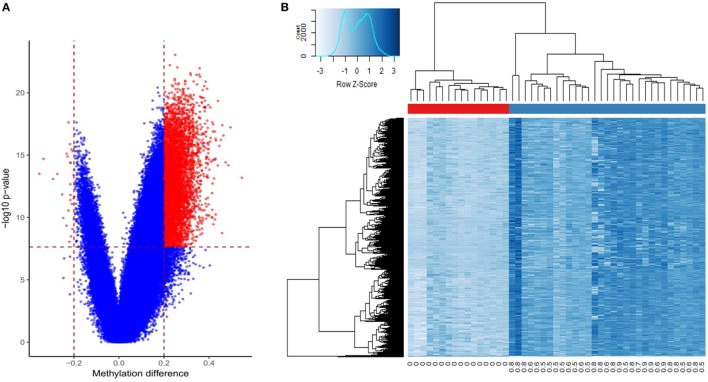
Methods: Archival PCa and normal tissues were assessed using genomic DNA methylation arrays. Differentially methylated sites and regions (DMRs) were used for functional assessment, gene-set enrichment and protein interaction analyses, and examination of transcription factor-binding patterns. Raw signal intensity data were used for identification of recurrent copy number variations (CNVs). Non-redundant fully differentiating cytosine-phosphate-guanine sites (CpGs), which did not overlap CNV segments, were used in an L1 regularized logistic regression model (LASSO) to train a classification algorithm. Validation of this algorithm was performed using a large external cohort of benign and tumor prostate arrays.
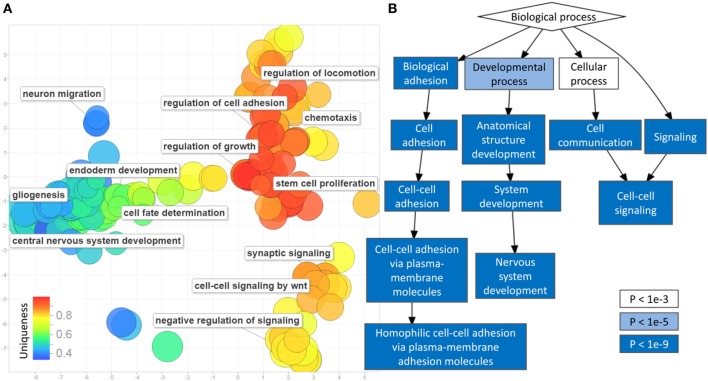
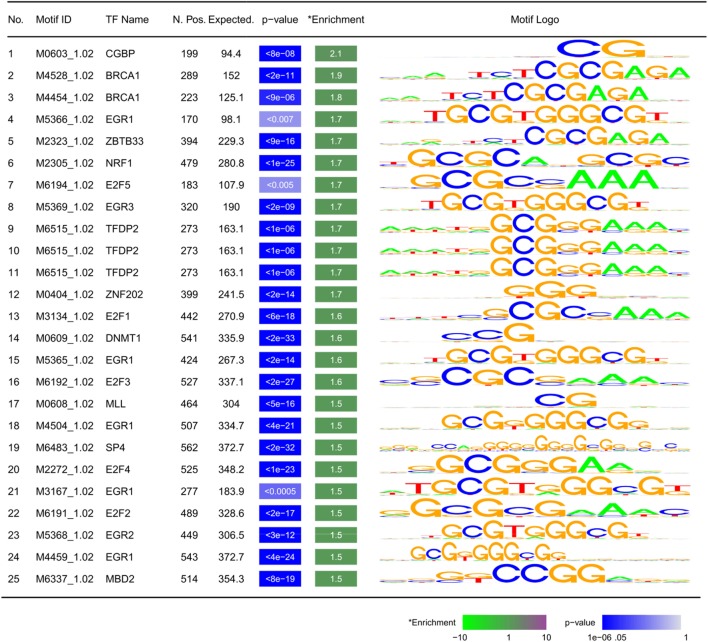
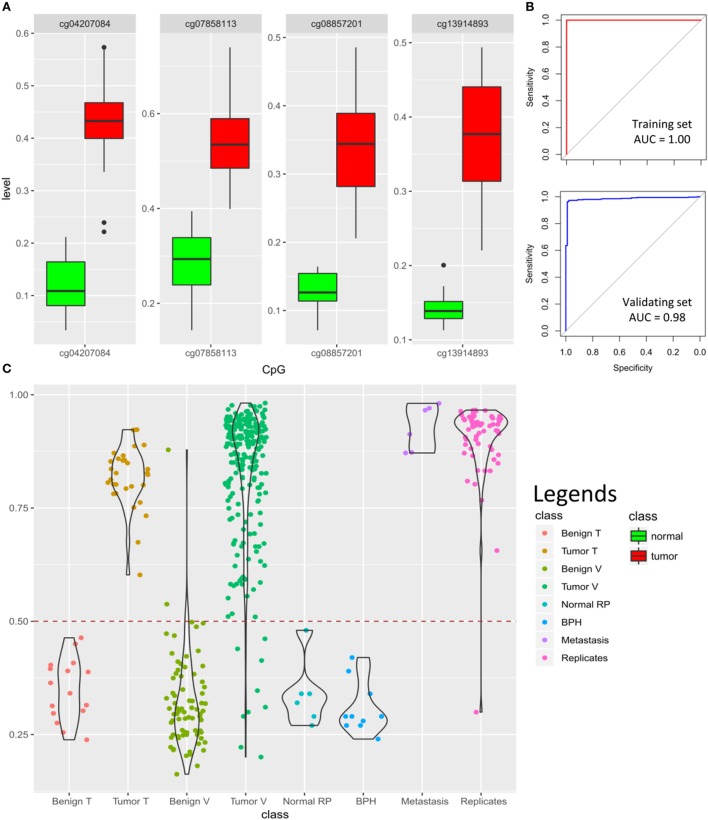
Results: Approximately 6,000 probes and 600 genomic regions showed significant DNA methylation changes, primarily involving hypermethylation. Gene-set enrichment and protein interaction analyses found an overrepresentation of genes related to cell communications, neurogenesis, and proliferation. Motif enrichment analysis demonstrated enrichment of tumor suppressor-binding sites nearby DMRs. Several of these regions were also found to contain copy number amplifications. Using four non-redundant fully differentiating CpGs, we trained a classification model with 100% accuracy in discriminating tumors from benign samples. Validation of this algorithm using an external cohort of 234 tumors and 92 benign samples yielded 96% sensitivity and 98% specificity. The model was found to be highly sensitive to detect metastatic lesions in bone, lymph node, and soft tissue, while being specific enough to differentiate the benign hyperplasia of prostate from tumor.
Conclusion: A considerable component of PCa DNA methylation profile represent driver events potentially established/maintained by disruption of tumor suppressor activity. As few as four CpGs from this profile can be used for screening of PCa.
Keywords: DNA methylation; LASSO; copy number variation; differentially methylated regions; machine learning; prostate cancer; protein interaction; transcription factor binding.
Figures

References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases