

De novo mutations in MED13, a component of the Mediator complex, are associated with a novel neurodevelopmental disorder
- PMID: 29740699
- PMCID: PMC5973976
- DOI: 10.1007/s00439-018-1887-y
De novo mutations in MED13, a component of the Mediator complex, are associated with a novel neurodevelopmental disorder
Abstract
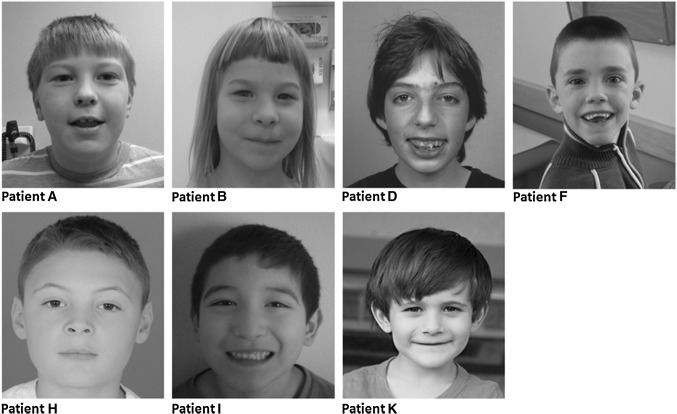
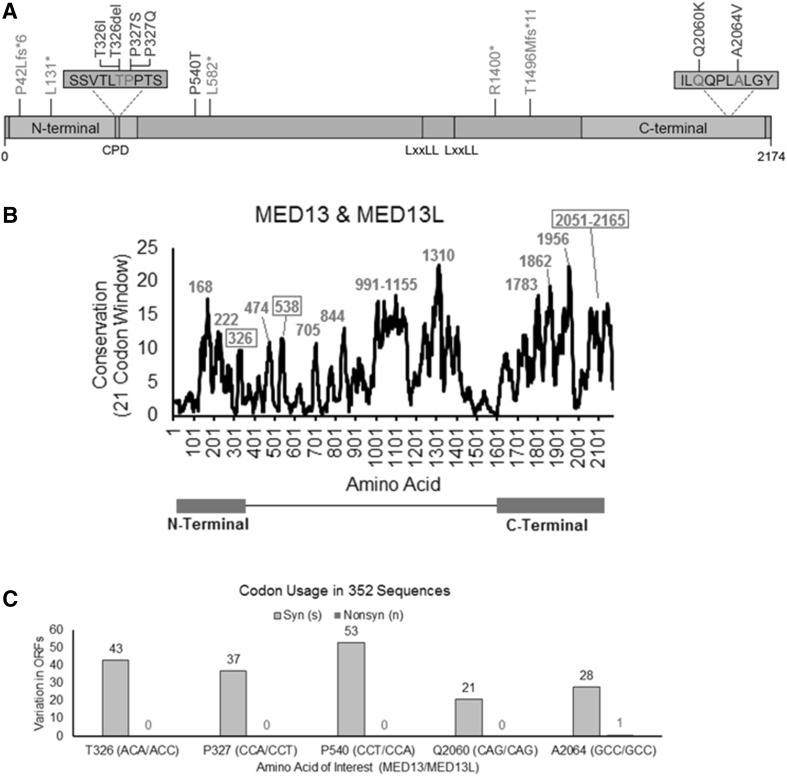
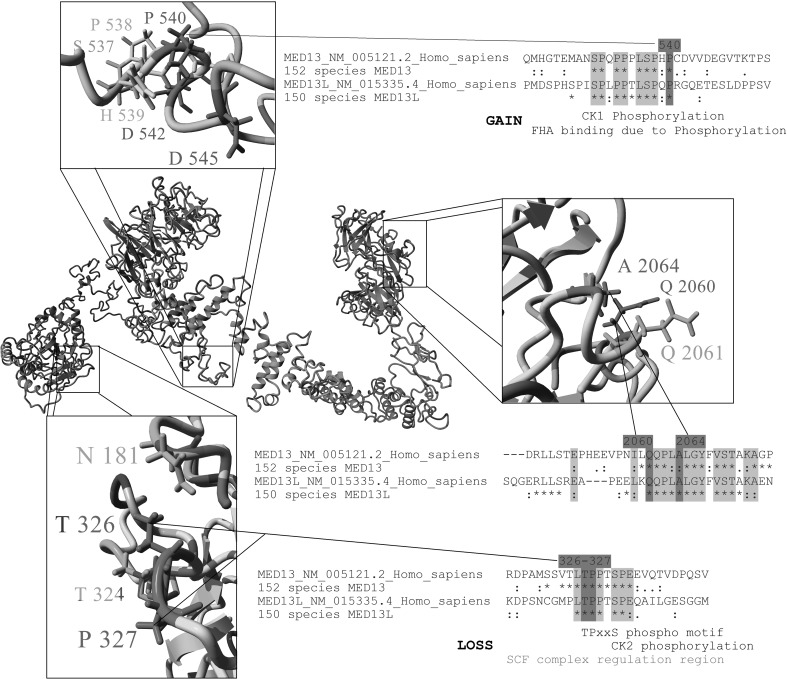
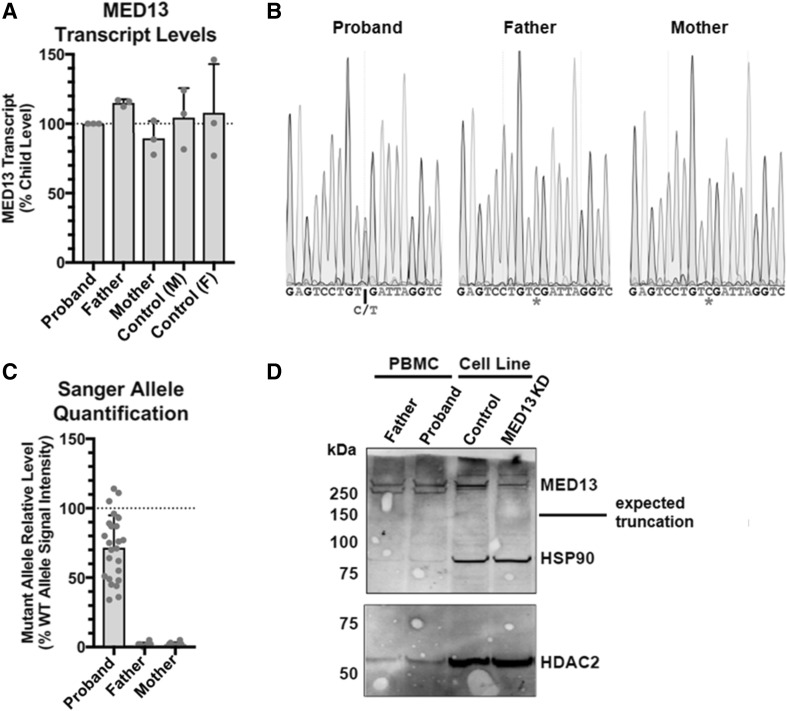
Many genetic causes of developmental delay and/or intellectual disability (DD/ID) are extremely rare, and robust discovery of these requires both large-scale DNA sequencing and data sharing. Here we describe a GeneMatcher collaboration which led to a cohort of 13 affected individuals harboring protein-altering variants, 11 of which are de novo, in MED13; the only inherited variant was transmitted to an affected child from an affected mother. All patients had intellectual disability and/or developmental delays, including speech delays or disorders. Other features that were reported in two or more patients include autism spectrum disorder, attention deficit hyperactivity disorder, optic nerve abnormalities, Duane anomaly, hypotonia, mild congenital heart abnormalities, and dysmorphisms. Six affected individuals had mutations that are predicted to truncate the MED13 protein, six had missense mutations, and one had an in-frame-deletion of one amino acid. Out of the seven non-truncating mutations, six clustered in two specific locations of the MED13 protein: an N-terminal and C-terminal region. The four N-terminal clustering mutations affect two adjacent amino acids that are known to be involved in MED13 ubiquitination and degradation, p.Thr326 and p.Pro327. MED13 is a component of the CDK8-kinase module that can reversibly bind Mediator, a multi-protein complex that is required for Polymerase II transcription initiation. Mutations in several other genes encoding subunits of Mediator have been previously shown to associate with DD/ID, including MED13L, a paralog of MED13. Thus, our findings add MED13 to the group of CDK8-kinase module-associated disease genes.
Conflict of interest statement
Conflict of interest
H.M., R.P. and A.C. are employees of GeneDx, Inc., a wholly owned subsidiary of OPKO Health, Inc. The other authors declare no conflict of interest.
Research involving human participants
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study. Additional informed consent was obtained from all individual participants for whom identifying information is included in this article.
Figures
References
-
- Andrews CV, Hunter DG, Engle EC (1993) Duane Syndrome. In: Pagon RA et al (eds) GeneReviews(R). Seattle (WA) - PubMed
-
- Au PYB, et al. GeneMatcher aids in the identification of a new malformation syndrome with intellectual disability, unique facial dysmorphisms, and skeletal and connective tissue abnormalities caused by de novo variants in. HNRNPK Hum Mut. 2015;36:1009–1014. doi: 10.1002/humu.22837. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
