

Astrocytes, an active player in Aicardi-Goutières syndrome
- PMID: 29740948
- PMCID: PMC8028286
- DOI: 10.1111/bpa.12600
Astrocytes, an active player in Aicardi-Goutières syndrome
Abstract
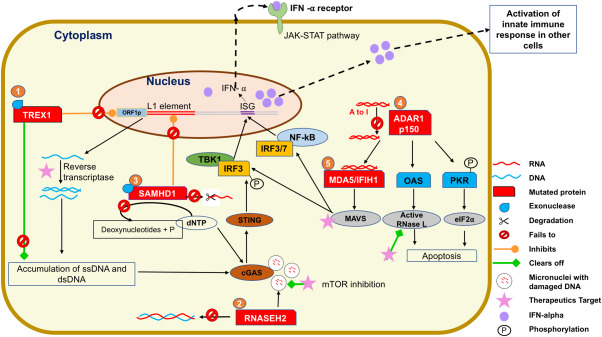
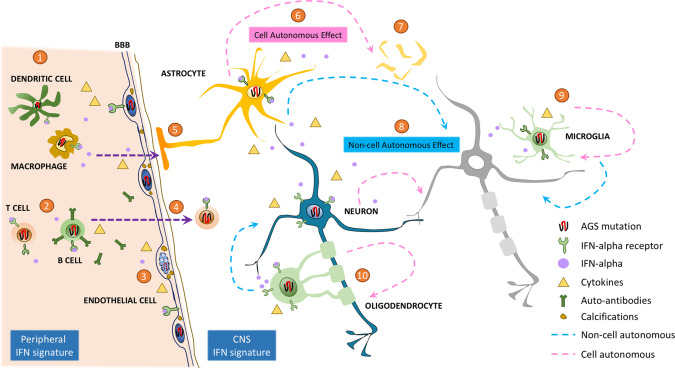
Aicardi-Goutières syndrome (AGS) is an early-onset, autoimmune and genetically heterogeneous disorder with severe neurologic injury. Molecular studies have established that autosomal recessive mutations in one of the following genes are causative: TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1 and IFIH1/MDA5. The phenotypic presentation and pathophysiology of AGS is associated with over-production of the cytokine Interferon-alpha (IFN-α) and its downstream signaling, characterized as type I interferonopathy. Astrocytes are one of the major source of IFN in the central nervous system (CNS) and it is proposed that they could be key players in AGS pathology. Astrocytes are the most ubiquitous glial cell in the CNS and perform a number of crucial and complex functions ranging from formation of blood-brain barrier, maintaining ionic homeostasis, metabolic support to synapse formation and elimination in healthy CNS. Involvement of astrocytic dysfunction in neurological diseases-Alexander's disease, Epilepsy, Alzheimer's and amyotrophic lateral sclerosis (ALS)-has been well-established. It is now known that compromised astrocytic function can contribute to CNS abnormalities and severe neurodegeneration, nevertheless, its contribution in AGS is unclear. The current review discusses known molecular and cellular pathways for AGS mutations and how it stimulates IFN-α signaling. We shed light on how astrocytes might be key players in the phenotypic presentations of AGS and emphasize the cell-autonomous and non-cell-autonomous role of astrocytes. Understanding the contribution of astrocytes will help reveal mechanisms underlying interferonopathy and develop targeted astrocyte specific therapeutic treatments in AGS.
Keywords: Aicardi-Goutières syndrome; astrocytes; interferon; type I interferonopathy.
© 2018 International Society of Neuropathology.
Figures
Similar articles
-
Aicardi-Goutières syndrome: A monogenic type I interferonopathy.Scand J Immunol. 2023 Oct;98(4):e13314. doi: 10.1111/sji.13314. Epub 2023 Jul 29. Scand J Immunol. 2023. PMID: 37515439 Review.
-
Phenotypic variation in Aicardi-Goutières syndrome explained by cell-specific IFN-stimulated gene response and cytokine release.J Immunol. 2015 Apr 15;194(8):3623-33. doi: 10.4049/jimmunol.1401334. Epub 2015 Mar 13. J Immunol. 2015. PMID: 25769924
-
Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutières Syndrome and Beyond.Neuropediatrics. 2016 Dec;47(6):355-360. doi: 10.1055/s-0036-1592307. Epub 2016 Sep 19. Neuropediatrics. 2016. PMID: 27643693 Review.
-
Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study.Lancet Neurol. 2013 Dec;12(12):1159-69. doi: 10.1016/S1474-4422(13)70258-8. Epub 2013 Oct 30. Lancet Neurol. 2013. PMID: 24183309 Free PMC article.
-
Aicardi-Goutières syndrome-associated mutation at ADAR1 gene locus activates innate immune response in mouse brain.J Neuroinflammation. 2021 Jul 31;18(1):169. doi: 10.1186/s12974-021-02217-9. J Neuroinflammation. 2021. PMID: 34332594 Free PMC article.
Cited by
-
Type I interferon (IFN)-inducible Absent in Melanoma 2 proteins in neuroinflammation: implications for Alzheimer's disease.J Neuroinflammation. 2019 Nov 26;16(1):236. doi: 10.1186/s12974-019-1639-5. J Neuroinflammation. 2019. PMID: 31771614 Free PMC article. Review.
-
Regulation of axon pathfinding by astroglia across genetic model organisms.Front Cell Neurosci. 2023 Oct 24;17:1241957. doi: 10.3389/fncel.2023.1241957. eCollection 2023. Front Cell Neurosci. 2023. PMID: 37941606 Free PMC article. Review.
-
The neurovascular unit in leukodystrophies: towards solving the puzzle.Fluids Barriers CNS. 2022 Feb 28;19(1):18. doi: 10.1186/s12987-022-00316-0. Fluids Barriers CNS. 2022. PMID: 35227276 Free PMC article. Review.
-
Pharmacological evaluation of drug therapies in Aicardi-Goutières syndrome: insights from patient-derived neural stem cells.Front Pharmacol. 2025 Mar 20;16:1549183. doi: 10.3389/fphar.2025.1549183. eCollection 2025. Front Pharmacol. 2025. PMID: 40183101 Free PMC article.
-
Preimplantation genetic testing for Aicardi-Goutières syndrome induced by novel compound heterozygous mutations of TREX1: an unaffected live birth.Mol Cytogenet. 2023 Jun 5;16(1):9. doi: 10.1186/s13039-023-00641-5. Mol Cytogenet. 2023. PMID: 37277873 Free PMC article.
References
-
- Aicardi J, Goutieres F (1984) A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol 15:49–54. - PubMed
-
- Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H et al (1998) Transgenic expression of IFN‐alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol 161:5016–5026. - PubMed
-
- Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, Aden K et al (2017) Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet 26:3960–3972. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
