

Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib
- PMID: 29743983
- PMCID: PMC5878876
- DOI: 10.1155/2018/7582730
Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib
Erratum in
-
Corrigendum to "Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib".Oxid Med Cell Longev. 2019 Jun 26;2019:9601435. doi: 10.1155/2019/9601435. eCollection 2019. Oxid Med Cell Longev. 2019. PMID: 31346363 Free PMC article.
Abstract
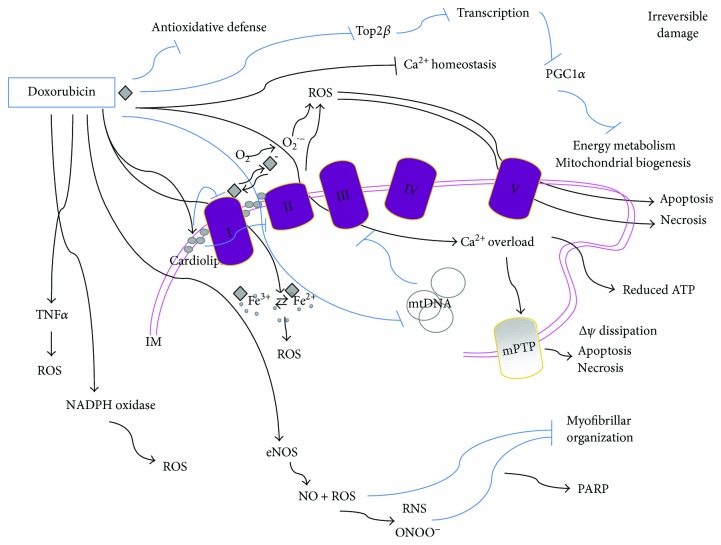
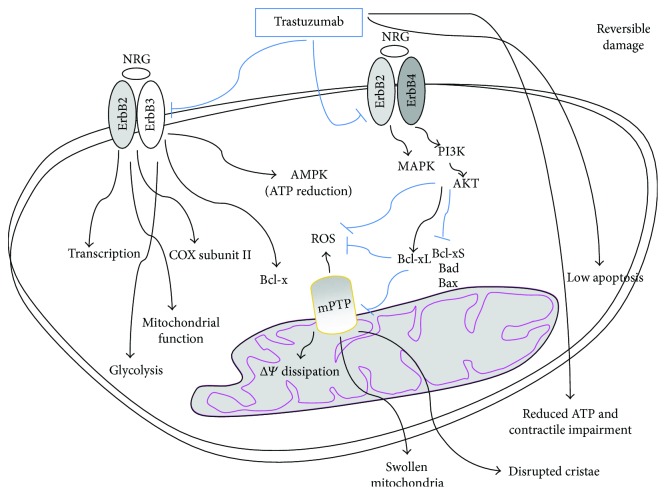
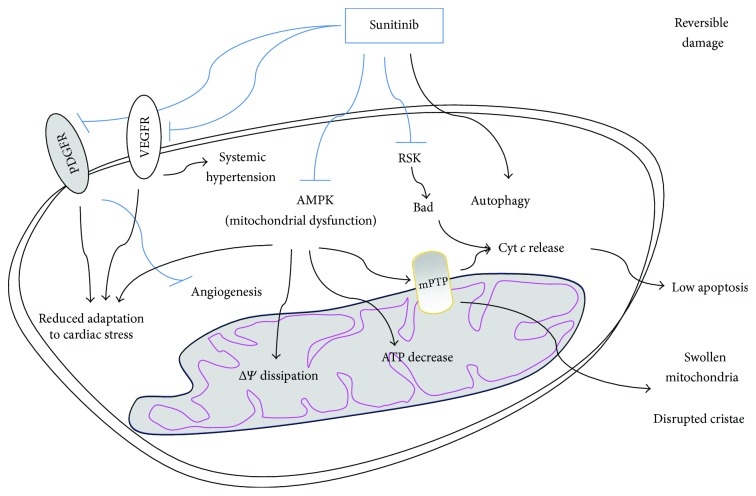
Many cancer therapies produce toxic side effects whose molecular mechanisms await full elucidation. The most feared and studied side effect of chemotherapeutic drugs is cardiotoxicity. Also, skeletal muscle physiology impairment has been recorded after many chemotherapeutical treatments. However, only doxorubicin has been extensively studied for its side effects on skeletal muscle. Chemotherapeutic-induced adverse side effects are, in many cases, mediated by mitochondrial damage. In particular, trastuzumab and sunitinib toxicity is mainly associated with mitochondria impairment and is mostly reversible. Vice versa, doxorubicin-induced toxicity not only includes mitochondria damage but can also lead to a more robust and extensive cell injury which is often irreversible and lethal. Drugs interfering with mitochondrial functionality determine the depletion of ATP reservoirs and lead to subsequent reversible contractile dysfunction. Mitochondrial damage includes the impairment of the respiratory chain and the loss of mitochondrial membrane potential with subsequent disruption of cellular energetic. In a context of increased stress, AMPK has a key role in maintaining energy homeostasis, and inhibition of the AMPK pathway is one of the proposed mechanisms possibly mediating mitochondrial toxicity due to chemotherapeutics. Therapies targeting and protecting cell metabolism and energy management might be useful tools in protecting muscular tissues against the toxicity induced by chemotherapeutic drugs.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
