

Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance
- PMID: 29748565
- PMCID: PMC5945626
- DOI: 10.1038/s41467-018-03917-2
Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance
Abstract
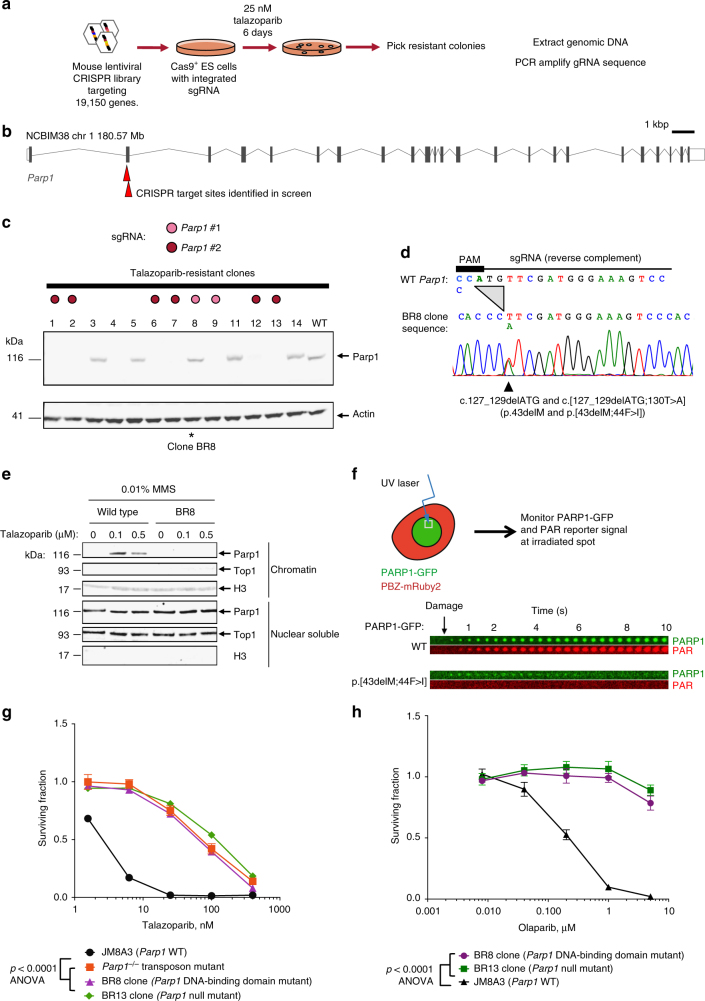
Although PARP inhibitors (PARPi) target homologous recombination defective tumours, drug resistance frequently emerges, often via poorly understood mechanisms. Here, using genome-wide and high-density CRISPR-Cas9 "tag-mutate-enrich" mutagenesis screens, we identify close to full-length mutant forms of PARP1 that cause in vitro and in vivo PARPi resistance. Mutations both within and outside of the PARP1 DNA-binding zinc-finger domains cause PARPi resistance and alter PARP1 trapping, as does a PARP1 mutation found in a clinical case of PARPi resistance. This reinforces the importance of trapped PARP1 as a cytotoxic DNA lesion and suggests that PARP1 intramolecular interactions might influence PARPi-mediated cytotoxicity. PARP1 mutations are also tolerated in cells with a pathogenic BRCA1 mutation where they result in distinct sensitivities to chemotherapeutic drugs compared to other mechanisms of PARPi resistance (BRCA1 reversion, 53BP1, REV7 (MAD2L2) mutation), suggesting that the underlying mechanism of PARPi resistance that emerges could influence the success of subsequent therapies.
Conflict of interest statement
C.J.L. and A.A. are named inventors on patents describing the use of PARP inhibitors and stand to gain from their development as part of the ICR “Rewards to Inventors” scheme. The remaining authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
