

Gene-by-environment interactions that disrupt mitochondrial homeostasis cause neurodegeneration in C. elegans Parkinson's models
- PMID: 29748634
- PMCID: PMC5945629
- DOI: 10.1038/s41419-018-0619-5
Gene-by-environment interactions that disrupt mitochondrial homeostasis cause neurodegeneration in C. elegans Parkinson's models
Abstract
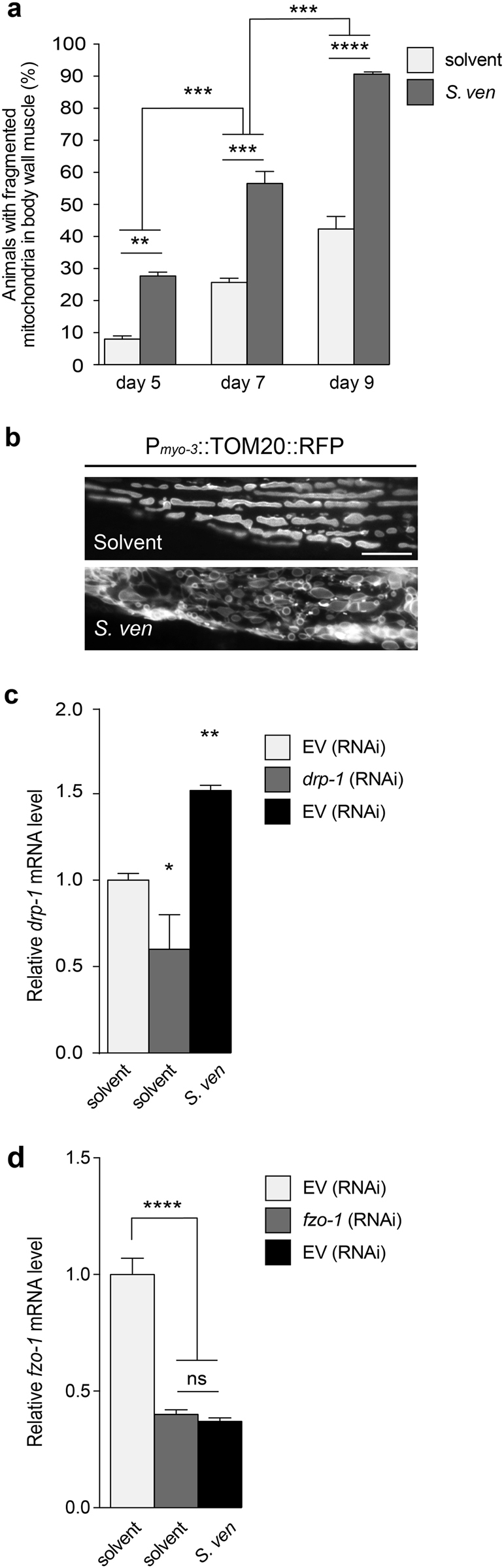
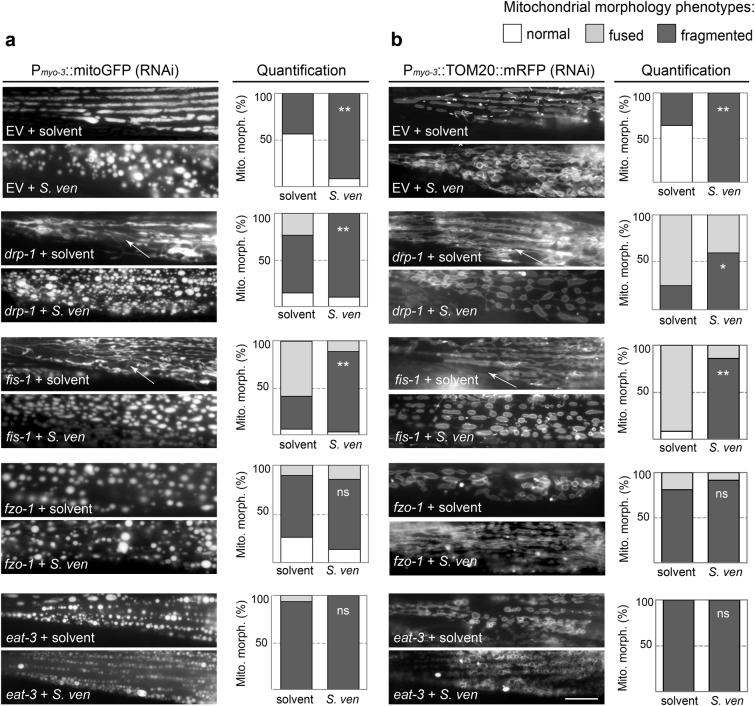
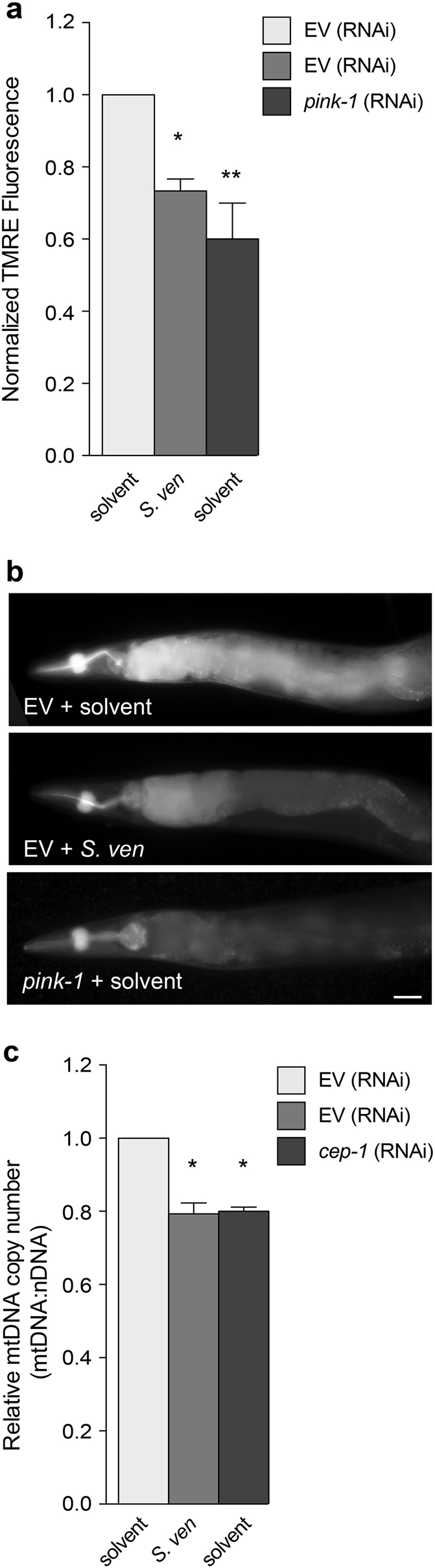
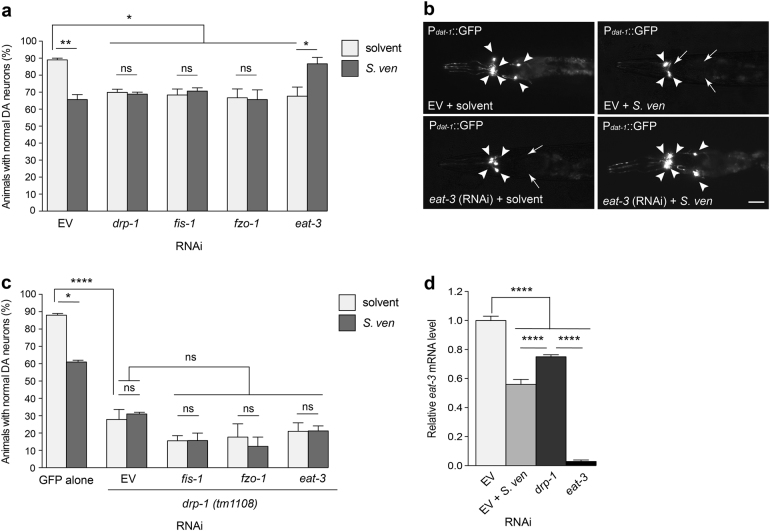
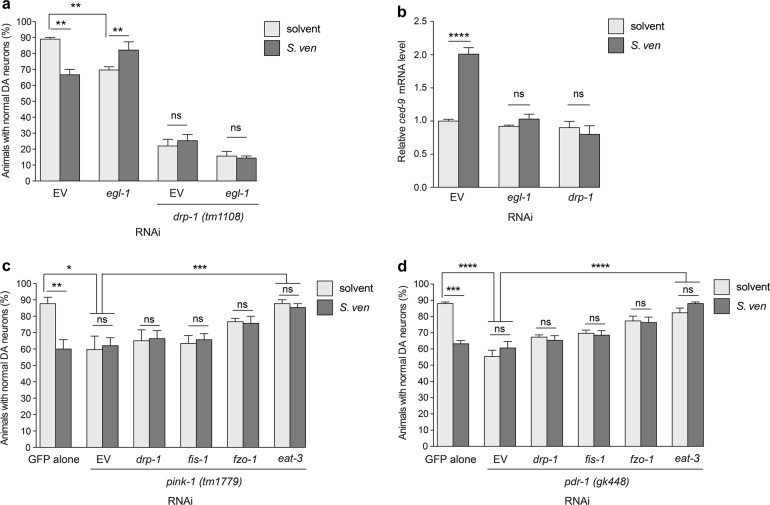
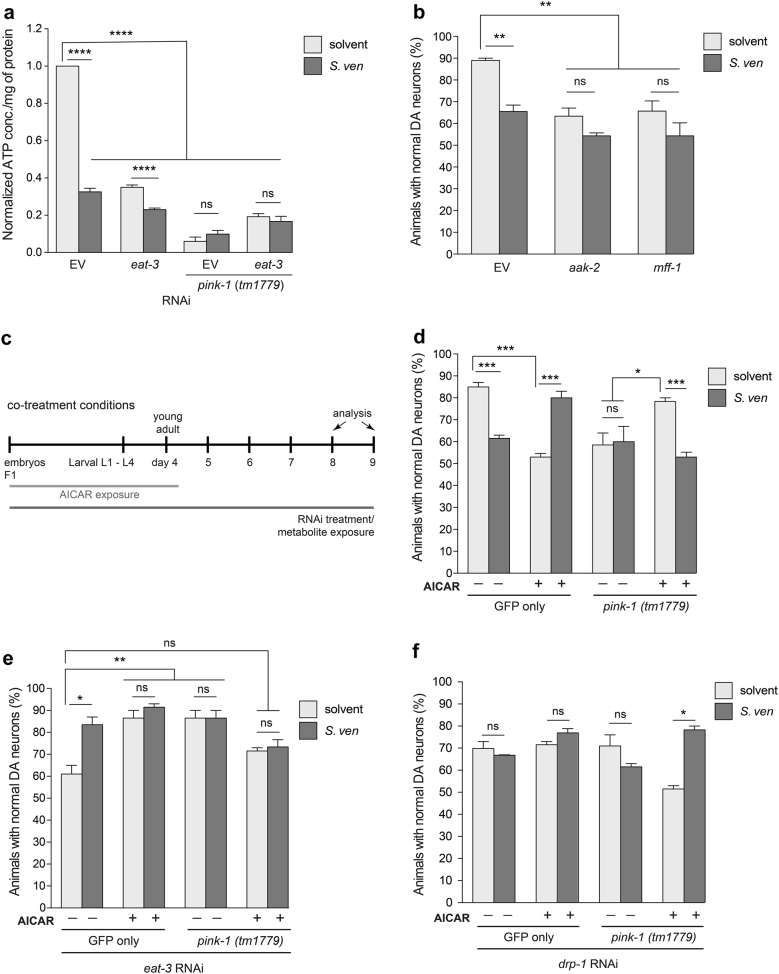
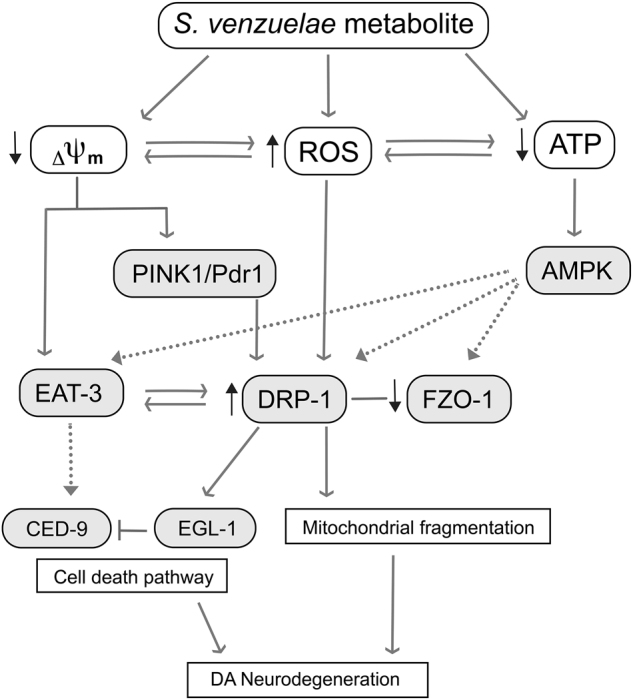
Parkinson's disease (PD) is a complex multifactorial disorder where environmental factors interact with genetic susceptibility. Accumulating evidence suggests that mitochondria have a central role in the progression of neurodegeneration in sporadic and/or genetic forms of PD. We previously reported that exposure to a secondary metabolite from the soil bacterium, Streptomyces venezuelae, results in age- and dose-dependent dopaminergic (DA) neurodegeneration in Caenorhabditis elegans and human SH-SY5Y neurons. Initial characterization of this environmental factor indicated that neurodegeneration occurs through a combination of oxidative stress, mitochondrial complex I impairment, and proteostatic disruption. Here we present extended evidence to elucidate the interaction between this bacterial metabolite and mitochondrial dysfunction in the development of DA neurodegeneration. We demonstrate that it causes a time-dependent increase in mitochondrial fragmentation through concomitant changes in the gene expression of mitochondrial fission and fusion components. In particular, the outer mitochondrial membrane fission and fusion genes, drp-1 (a dynamin-related GTPase) and fzo-1 (a mitofusin homolog), are up- and down-regulated, respectively. Additionally, eat-3, an inner mitochondrial membrane fusion component, an OPA1 homolog, is also down regulated. These changes are associated with a metabolite-induced decline in mitochondrial membrane potential and enhanced DA neurodegeneration that is dependent on PINK-1 function. Genetic analysis also indicates an association between the cell death pathway and drp-1 following S. ven exposure. Metabolite-induced neurotoxicity can be suppressed by DA-neuron-specific RNAi knockdown of eat-3. AMPK activation by 5-amino-4-imidazole carboxamide riboside (AICAR) ameliorated metabolite- or PINK-1-induced neurotoxicity; however, it enhanced neurotoxicity under normal conditions. These studies underscore the critical role of mitochondrial dynamics in DA neurodegeneration. Moreover, given the largely undefined environmental components of PD etiology, these results highlight a response to an environmental factor that defines distinct mechanisms underlying a potential contributor to the progressive DA neurodegeneration observed in PD.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Mitochondrial dysfunction, oxidative stress, and neurodegeneration elicited by a bacterial metabolite in a C. elegans Parkinson's model.Cell Death Dis. 2014 Jan 9;5(1):e984. doi: 10.1038/cddis.2013.513. Cell Death Dis. 2014. PMID: 24407237 Free PMC article.
-
Zearalenone Induces Dopaminergic Neurodegeneration via DRP-1-Involved Mitochondrial Fragmentation and Apoptosis in a Caenorhabditis elegans Parkinson's Disease Model.J Agric Food Chem. 2021 Oct 13;69(40):12030-12038. doi: 10.1021/acs.jafc.1c05836. Epub 2021 Sep 29. J Agric Food Chem. 2021. PMID: 34586801
-
Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. elegans Models of Parkinson's Disease.J Neurosci. 2017 Nov 15;37(46):11085-11100. doi: 10.1523/JNEUROSCI.1294-17.2017. Epub 2017 Oct 13. J Neurosci. 2017. PMID: 29030433 Free PMC article.
-
No Country for Old Worms: A Systematic Review of the Application of C. elegans to Investigate a Bacterial Source of Environmental Neurotoxicity in Parkinson's Disease.Metabolites. 2018 Oct 29;8(4):70. doi: 10.3390/metabo8040070. Metabolites. 2018. PMID: 30380609 Free PMC article. Review.
-
Exploring Caenorhabditis elegans as Parkinson's Disease Model: Neurotoxins and Genetic Implications.Neurotox Res. 2024 Feb 6;42(1):11. doi: 10.1007/s12640-024-00686-3. Neurotox Res. 2024. PMID: 38319410 Review.
Cited by
-
Alcohol induces mitochondrial fragmentation and stress responses to maintain normal muscle function in Caenorhabditis elegans.FASEB J. 2020 Jun;34(6):8204-8216. doi: 10.1096/fj.201903166R. Epub 2020 Apr 15. FASEB J. 2020. PMID: 32294300 Free PMC article.
-
Aberrant Mitochondrial Dynamics: An Emerging Pathogenic Driver of Abdominal Aortic Aneurysm.Cardiovasc Ther. 2021 Jun 16;2021:6615400. doi: 10.1155/2021/6615400. eCollection 2021. Cardiovasc Ther. 2021. PMID: 34221126 Free PMC article. Review.
-
Xanthine Dehydrogenase Is a Modulator of Dopaminergic Neurodegeneration in Response to Bacterial Metabolite Exposure in C. elegans.Cells. 2023 Apr 15;12(8):1170. doi: 10.3390/cells12081170. Cells. 2023. PMID: 37190079 Free PMC article.
-
Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans.Cell Mol Life Sci. 2019 May;76(10):1967-1985. doi: 10.1007/s00018-019-03024-5. Epub 2019 Mar 6. Cell Mol Life Sci. 2019. PMID: 30840087 Free PMC article.
-
Gene-environment interactions in birth defect etiology: Challenges and opportunities.Curr Top Dev Biol. 2023;152:1-30. doi: 10.1016/bs.ctdb.2022.10.001. Epub 2022 Nov 14. Curr Top Dev Biol. 2023. PMID: 36707208 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
