

Genetic characteristics of patients with congenital hyperinsulinism
- PMID: 29750770
- PMCID: PMC6084463
- DOI: 10.1097/MOP.0000000000000645
Genetic characteristics of patients with congenital hyperinsulinism
Abstract
Purpose of review: Congenital hyperinsulinism is the most common cause of persistent hypoglycemia in infants and children. Early and appropriate recognition and treatment of hypoglycemia is vital to minimize neurocognitive impairment.
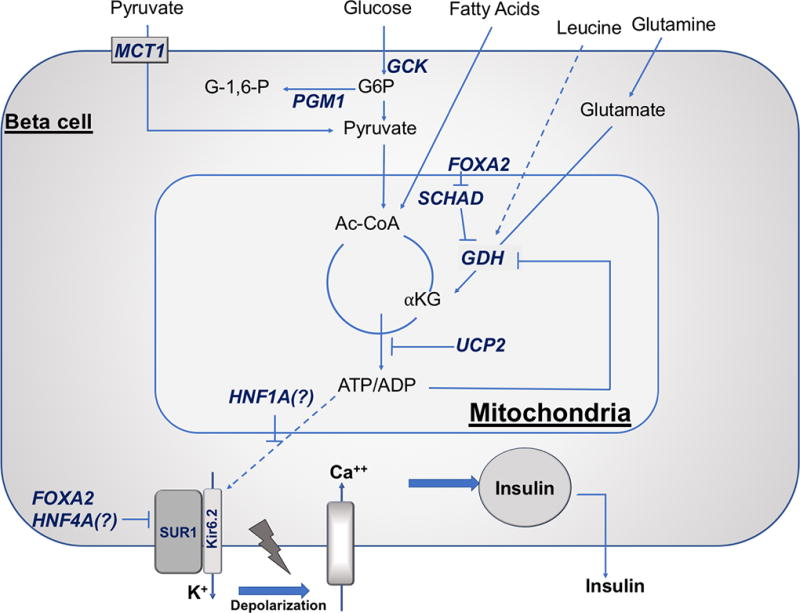
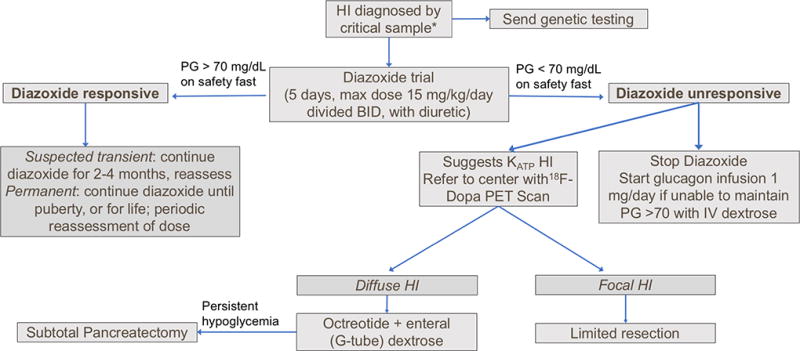
Recent findings: There are at least 11 known monogenic forms of hyperinsulinism and several associated syndromes. Molecular diagnosis allows for prediction of the effectiveness of diazoxide and the likelihood of focal hyperinsulinism. Inactivating mutations in the genes encoding the ATP-sensitive potassium channel (KATP hyperinsulinism) account for 60% of all identifiable mutations, including 85% of diazoxide-unresponsive cases. Syndromes or disorders associated with hyperinsulinism include Beckwith-Wiedemann syndrome, Kabuki syndrome, Turner syndrome, and congenital disorders of glycosylation. Although focal hyperinsulinism can be cured by resection of the lesion, therapeutic options for nonfocal hyperinsulinism remain limited and include diazoxide, octreotide, long-acting somatostatin analogs, and near-total pancreatectomy. Although sirolimus has been reported to improve glycemic control in infants with diazoxide-unresponsive hyperinsulinism, the extent of improvement has been limited, and significant adverse events have been reported.
Summary: Identification of the cause of congenital hyperinsulinism helps guide management decisions. Use of therapies with limited benefit and significant potential risks should be avoided.
Conflict of interest statement
The authors have no conflict of interest.
Figures
References
-
- Ludwig A, Enke S, Heindorf J, Empting S, Meissner T, Mohnike K. Formal Neurocognitive Testing in 60 Patients with Congenital Hyperinsulinism. Horm Res Paediatr. 2018;89(1):1–6. This prospective study of 60 patients with HI used formal neurocognitive testing to evaluate developmental delay. - PubMed
-
- Helleskov A, Melikyan M, Globa E, Shcherderkina I, Poertner F, Larsen AM, et al. Both Low Blood Glucose and Insufficient Treatment Confer Risk of Neurodevelopmental Impairment in Congenital Hyperinsulinism: A Multinational Cohort Study. Front Endocrinol (Lausanne) 2017;8:156. This report of 75 patients with HI from two European centers describes risk of neurodevelopmental impairment related to both severe hypoglycemia and treatment delay. - PMC - PubMed
-
- Ferrara C, Patel P, Becker S, Stanley CA, Kelly A. Biomarkers of Insulin for the Diagnosis of Hyperinsulinemic Hypoglycemia in Infants and Children. J Pediatr. 2016;168:212–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
