

Ion Channel Modulators in Cystic Fibrosis
- PMID: 29750923
- PMCID: PMC6113631
- DOI: 10.1016/j.chest.2018.04.036
Ion Channel Modulators in Cystic Fibrosis
Abstract
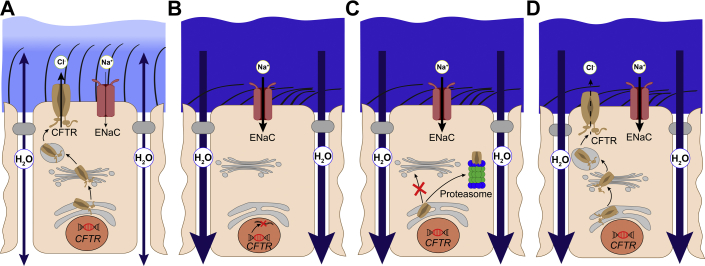
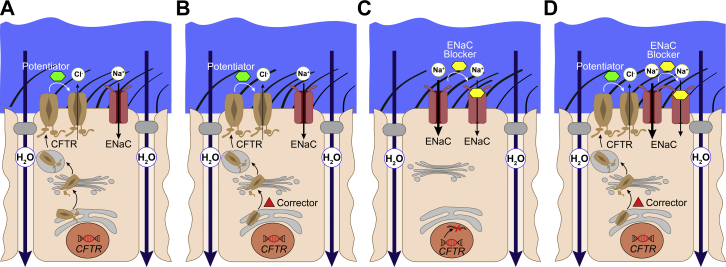
Cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and remains one of the most common life-shortening genetic diseases affecting the lung and other organs. CFTR functions as a cyclic adenosine monophosphate-dependent anion channel that transports chloride and bicarbonate across epithelial surfaces, and disruption of these ion transport processes plays a central role in the pathogenesis of CF. These findings provided the rationale for pharmacologic modulation of ion transport, either by targeting mutant CFTR or alternative ion channels that can compensate for CFTR dysfunction, as a promising therapeutic approach. High-throughput screening has supported the development of CFTR modulator compounds. CFTR correctors are designed to improve defective protein processing, trafficking, and cell surface expression, whereas potentiators increase the activity of mutant CFTR at the cell surface. The approval of the first potentiator ivacaftor for the treatment of patients with specific CFTR mutations and, more recently, the corrector lumacaftor in combination with ivacaftor for patients homozygous for the common F508del mutation, were major breakthroughs on the path to causal therapies for all patients with CF. The present review focuses on recent developments and remaining challenges of CFTR-directed therapies, as well as modulators of other ion channels such as alternative chloride channels and the epithelial sodium channel as additional targets in CF lung disease. We further discuss how patient-derived precision medicine models may aid the translation of emerging next-generation ion channel modulators from the laboratory to the clinic and tailor their use for optimal therapeutic benefits in individual patients with CF.
Keywords: cystic fibrosis; pharmacotherapy; translating basic research.
Copyright © 2018 American College of Chest Physicians. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Elborn J.S. Cystic fibrosis. Lancet. 2016;388(10059):2519–2531. - PubMed
-
- Mall M.A., Hartl D. CFTR: cystic fibrosis and beyond. Eur Respir J. 2014;44(4):1042–1054. - PubMed
-
- Cohen-Cymberknoh M., Shoseyov D., Kerem E. Managing cystic fibrosis: strategies that increase life expectancy and improve quality of life. Am J Respir Crit Care Med. 2011;183(11):1463–1471. - PubMed
-
- Stern M., Bertrand D.P., Bignamini E. European Cystic Fibrosis Society Standards of Care: quality management in cystic fibrosis. J Cyst Fibros. 2014;13(suppl 1):S43–S59. - PubMed
-
- Riordan J.R., Rommens J.M., Kerem B. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
