

Single-cell RNA-seq of human induced pluripotent stem cells reveals cellular heterogeneity and cell state transitions between subpopulations
- PMID: 29752298
- PMCID: PMC6028138
- DOI: 10.1101/gr.223925.117
Single-cell RNA-seq of human induced pluripotent stem cells reveals cellular heterogeneity and cell state transitions between subpopulations
Abstract
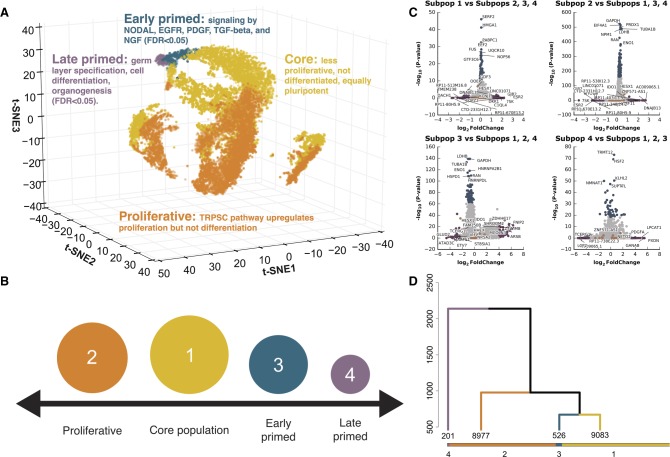
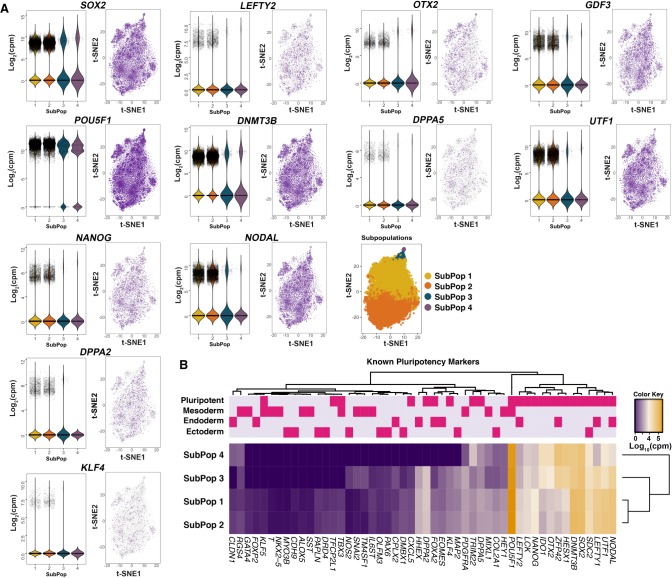
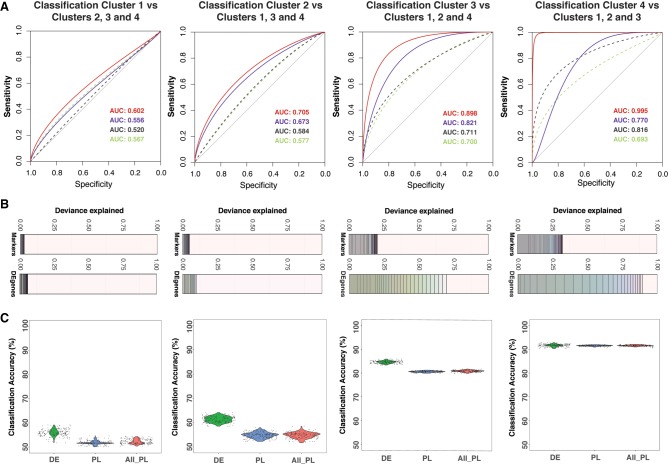
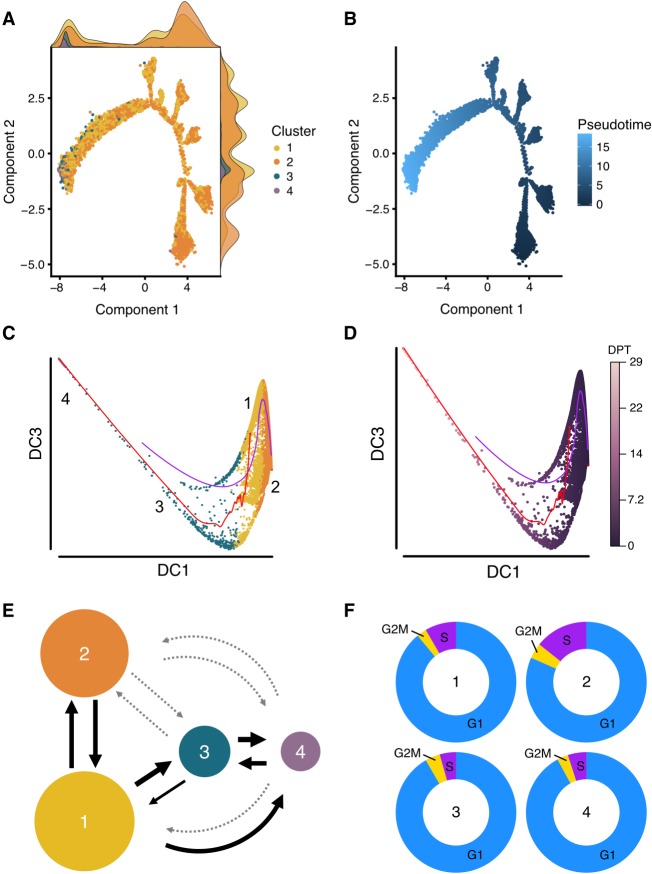
Heterogeneity of cell states represented in pluripotent cultures has not been described at the transcriptional level. Since gene expression is highly heterogeneous between cells, single-cell RNA sequencing can be used to identify how individual pluripotent cells function. Here, we present results from the analysis of single-cell RNA sequencing data from 18,787 individual WTC-CRISPRi human induced pluripotent stem cells. We developed an unsupervised clustering method and, through this, identified four subpopulations distinguishable on the basis of their pluripotent state, including a core pluripotent population (48.3%), proliferative (47.8%), early primed for differentiation (2.8%), and late primed for differentiation (1.1%). For each subpopulation, we were able to identify the genes and pathways that define differences in pluripotent cell states. Our method identified four transcriptionally distinct predictor gene sets composed of 165 unique genes that denote the specific pluripotency states; using these sets, we developed a multigenic machine learning prediction method to accurately classify single cells into each of the subpopulations. Compared against a set of established pluripotency markers, our method increases prediction accuracy by 10%, specificity by 20%, and explains a substantially larger proportion of deviance (up to threefold) from the prediction model. Finally, we developed an innovative method to predict cells transitioning between subpopulations and support our conclusions with results from two orthogonal pseudotime trajectory methods.
© 2018 Nguyen et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Angerer P, Haghverdi L, Büttner M, Theis FJ, Marr C, Buettner F. 2016. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32: 1241–1243. - PubMed
-
- Artavanis-Tsakonas S, Rand MD, Lake RJ. 1999. Notch signaling: cell fate control and signal integration in development. Science 284: 770–776. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources