

Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus α-synuclein and β-amyloid aggregates
- PMID: 29752409
- PMCID: PMC6036193
- DOI: 10.1074/jbc.RA117.000378
Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus α-synuclein and β-amyloid aggregates
Abstract
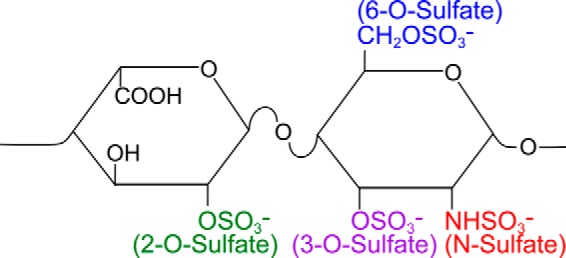
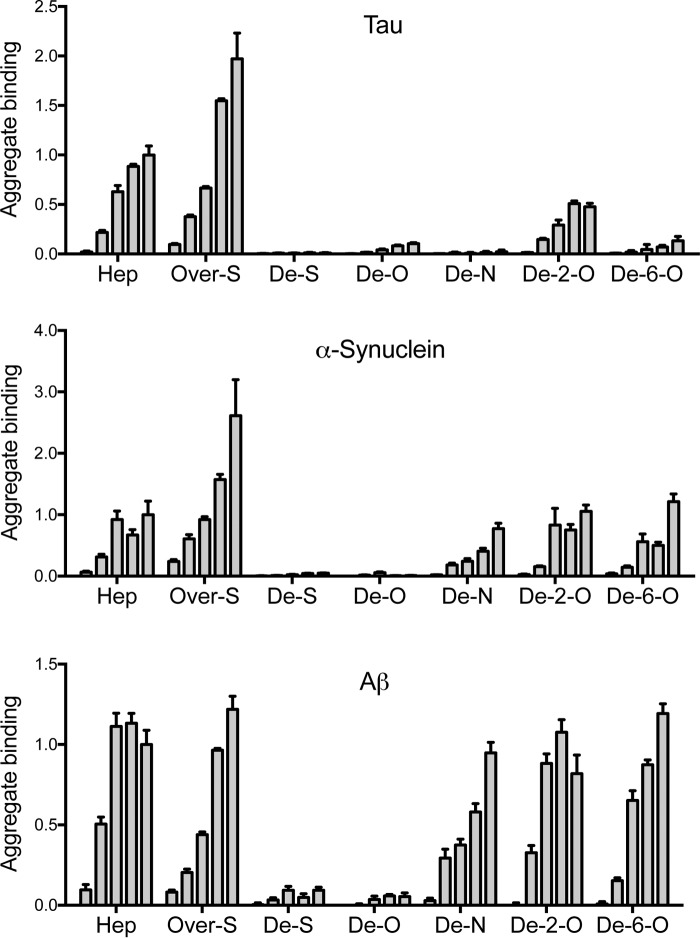
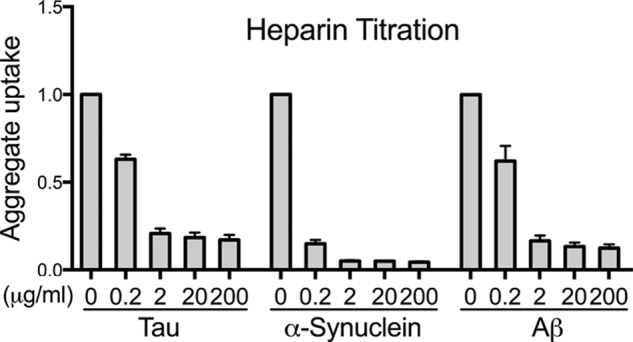
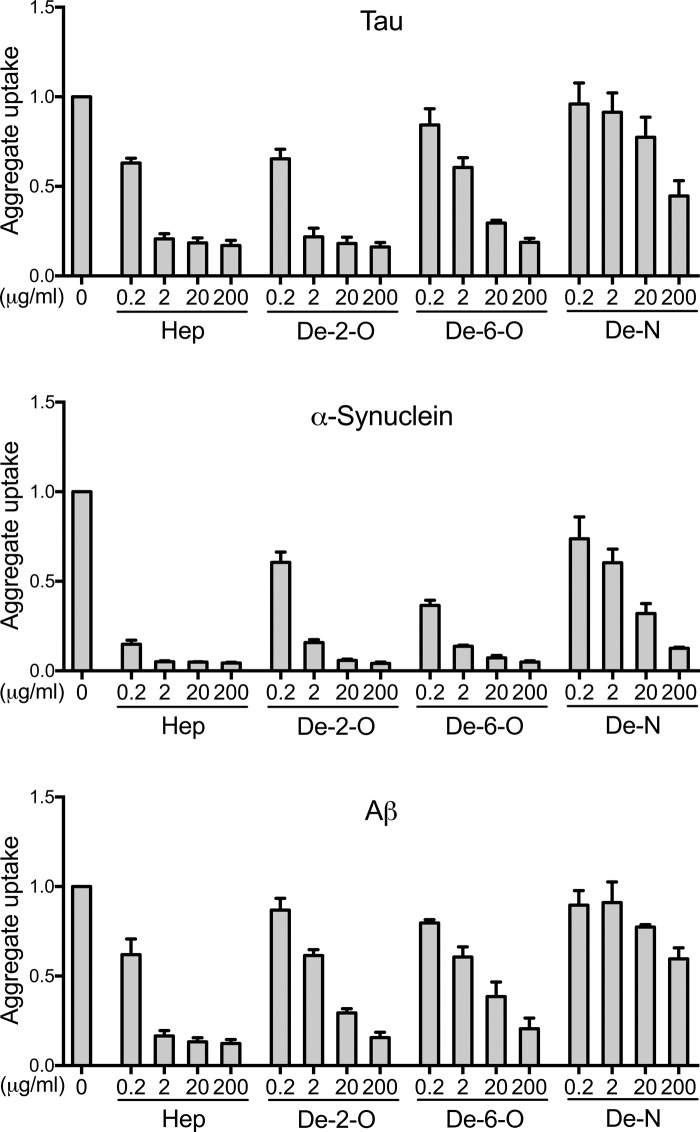
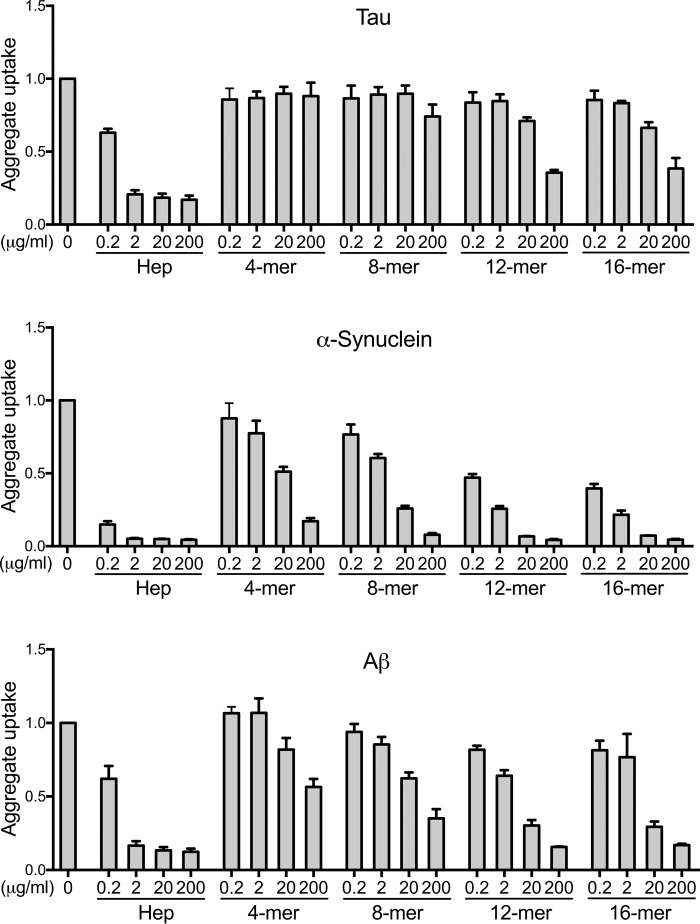
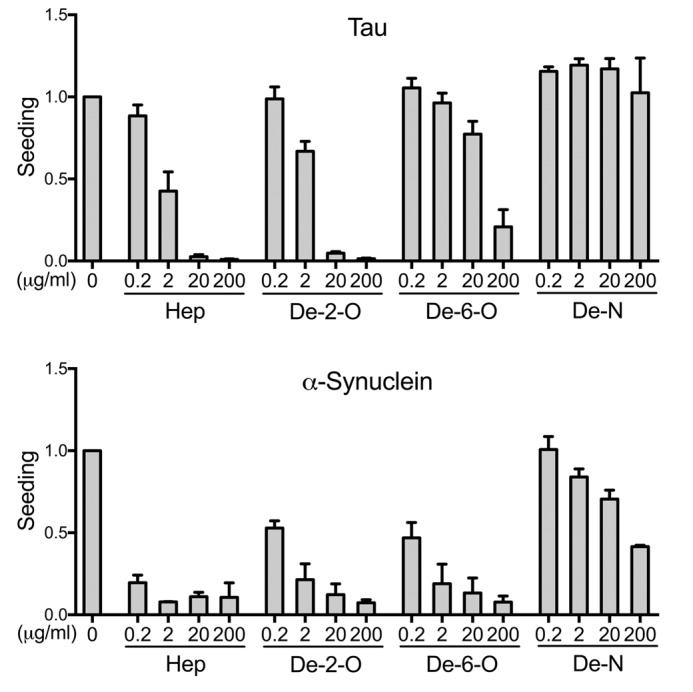
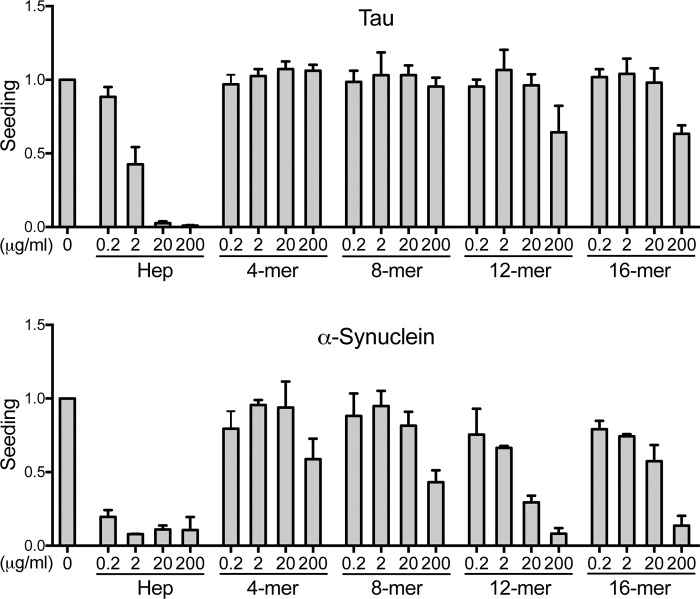
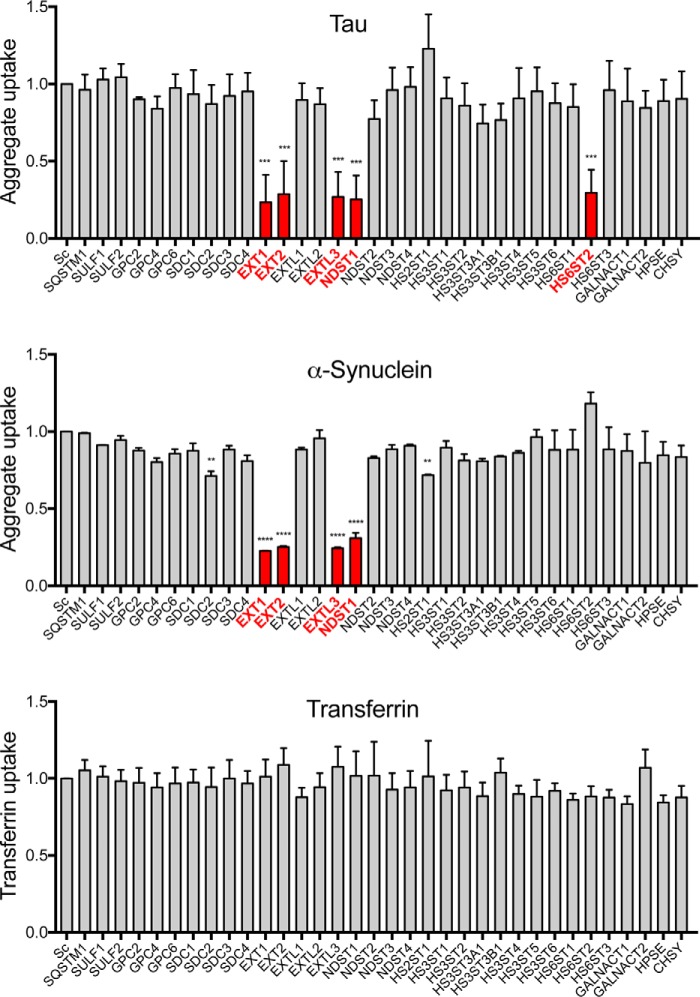
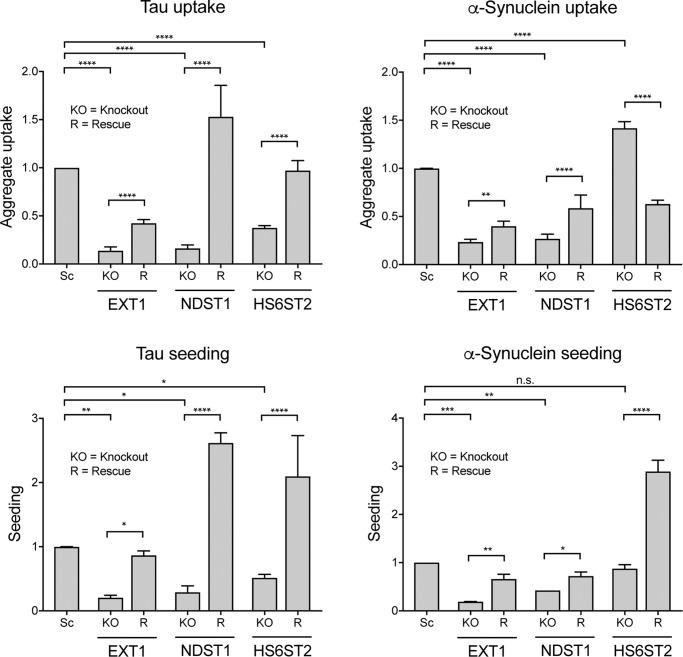
Transcellular propagation of protein aggregate "seeds" has been proposed to mediate the progression of neurodegenerative diseases in tauopathies and α-synucleinopathies. We previously reported that tau and α-synuclein aggregates bind heparan sulfate proteoglycans (HSPGs) on the cell surface, promoting cellular uptake and intracellular seeding. However, the specificity and binding mode of these protein aggregates to HSPGs remain unknown. Here, we measured direct interaction with modified heparins to determine the size and sulfation requirements for tau, α-synuclein, and β-amyloid (Aβ) aggregate binding to glycosaminoglycans (GAGs). Varying the GAG length and sulfation patterns, we next conducted competition studies with heparin derivatives in cell-based assays. Tau aggregates required a precise GAG architecture with defined sulfate moieties in the N- and 6-O-positions, whereas the binding of α-synuclein and Aβ aggregates was less stringent. To determine the genes required for aggregate uptake, we used CRISPR/Cas9 to individually knock out the major genes of the HSPG synthesis pathway in HEK293T cells. Knockouts of the extension enzymes exostosin 1 (EXT1), exostosin 2 (EXT2), and exostosin-like 3 (EXTL3), as well as N-sulfotransferase (NDST1) or 6-O-sulfotransferase (HS6ST2) significantly reduced tau uptake, consistent with our biochemical findings, and knockouts of EXT1, EXT2, EXTL3, or NDST1, but not HS6ST2 reduced α-synuclein uptake. In summary, tau aggregates display specific interactions with HSPGs that depend on GAG length and sulfate moiety position, whereas α-synuclein and Aβ aggregates exhibit more flexible interactions with HSPGs. These principles may inform the development of mechanism-based therapies to block transcellular propagation of amyloid protein-based pathologies.
Keywords: Alzheimer disease; CRISPR/Cas; HSPG; Tau protein (Tau); amyloid-β (Aβ); heparan sulfate; neurodegeneration; neurodegenerative disease; proteoglycan; tauopathies; α-synuclein.
© 2018 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Holmes B. B., DeVos S. L., Kfoury N., Li M., Jacks R., Yanamandra K., Ouidja M. O., Brodsky F. M., Marasa J., Bagchi D. P., Kotzbauer P. T., Miller T. M., Papy-Garcia D., and Diamond M. I. (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110, E3138–E3147 10.1073/pnas.1301440110 - DOI - PMC - PubMed
-
- Wang Z., Hsieh P. H., Xu Y., Thieker D., Chai E. J., Xie S., Cooley B., Woods R. J., Chi L., and Liu J. (2017) Synthesis of 3-O-sulfated oligosaccharides to understand the relationship between structures and functions of heparan sulfate. J. Am. Chem. Soc. 2017 10.1021/jacs.7b01923 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
