

Atomic Resolution Cryo-EM Structure of β-Galactosidase
- PMID: 29754826
- PMCID: PMC6129192
- DOI: 10.1016/j.str.2018.04.004
Atomic Resolution Cryo-EM Structure of β-Galactosidase
Abstract
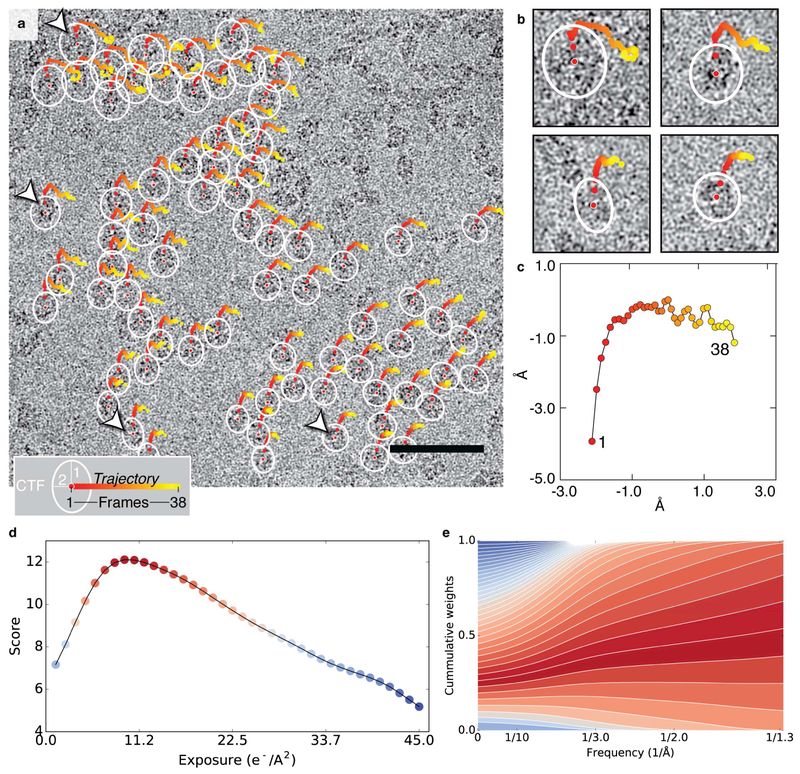
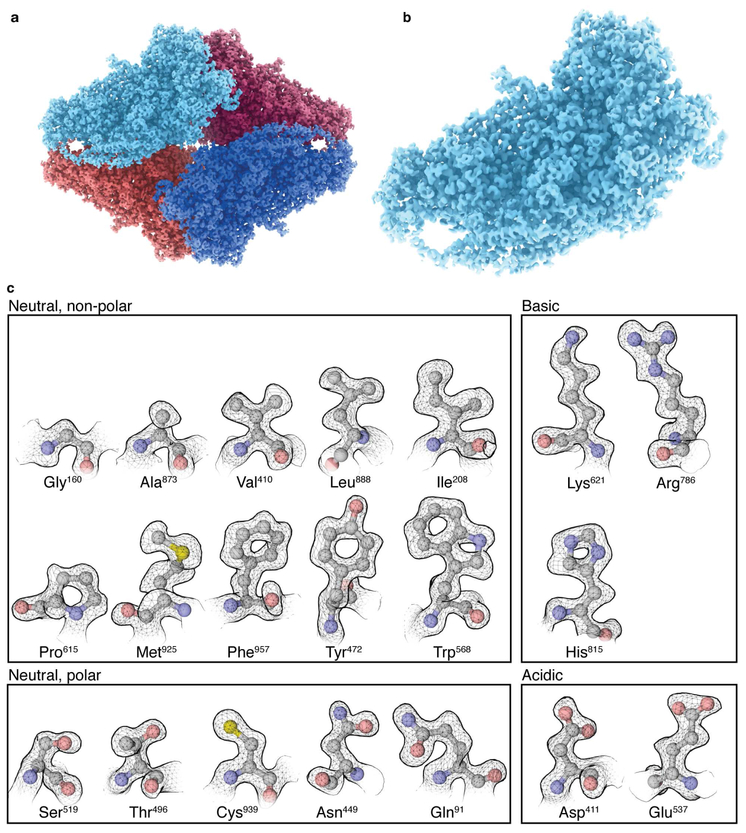
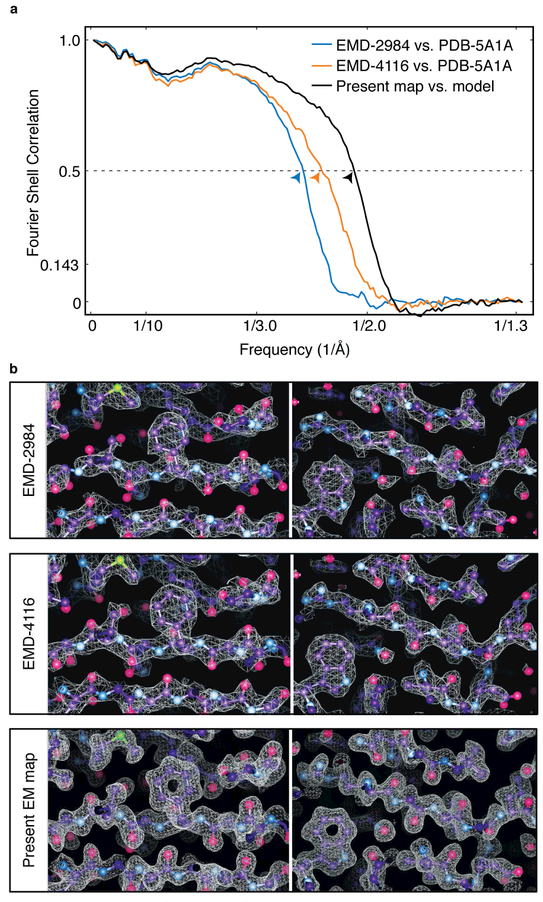
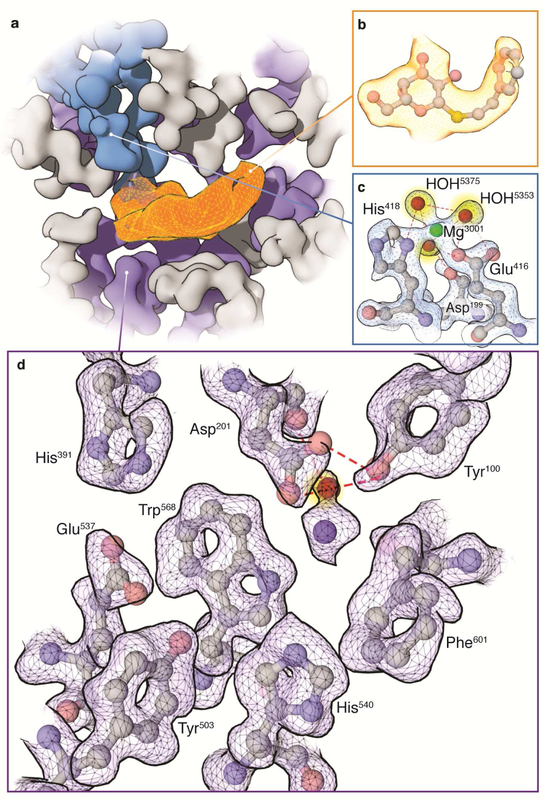
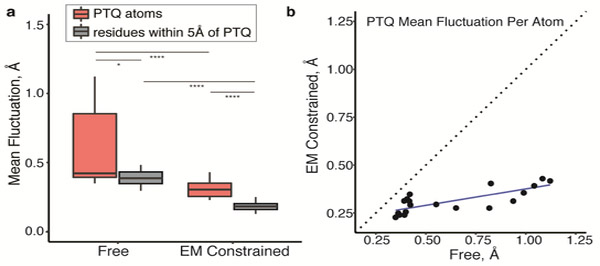
The advent of direct electron detectors has enabled the routine use of single-particle cryo-electron microscopy (EM) approaches to determine structures of a variety of protein complexes at near-atomic resolution. Here, we report the development of methods to account for local variations in defocus and beam-induced drift, and the implementation of a data-driven dose compensation scheme that significantly improves the extraction of high-resolution information recorded during exposure of the specimen to the electron beam. These advances enable determination of a cryo-EM density map for β-galactosidase bound to the inhibitor phenylethyl β-D-thiogalactopyranoside where the ordered regions are resolved at a level of detail seen in X-ray maps at ∼ 1.5 Å resolution. Using this density map in conjunction with constrained molecular dynamics simulations provides a measure of the local flexibility of the non-covalently bound inhibitor and offers further opportunities for structure-guided inhibitor design.
Keywords: atomic resolution; computer-aided drug discovery; drift correction; drug discovery; high-resolution protein structure; precision medicine; radiation damage; single-particle cryo-EM.
Published by Elsevier Ltd.
Figures
References
-
- Abrishami V, Vargas J, Li X, Cheng Y, Marabini R, Sorzano C, Oscar S, and Carazo JM (2015). Alignment of direct detection device micrographs using a robust Optical Flow approach. Journal of Structural Biology 189, 163–176. - PubMed
-
- Aguerrebere C, Delbracio M, Bartesaghi A, and Sapiro G (2016). Fundamental Limits in Multi-Image Alignment. IEEE Transactions on Signal Processing 64, 5707–5722.
-
- Baker LA, Smith EA, Bueler SA, and Rubinstein JL (2010). The resolution dependence of optimal exposures in liquid nitrogen temperature electron cryomicroscopy of catalase crystals. Journal of Structural Biology 169, 431–437. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
