

Analysis of Phylogenetic Variation of Stenotrophomonas maltophilia Reveals Human-Specific Branches
- PMID: 29755435
- PMCID: PMC5932162
- DOI: 10.3389/fmicb.2018.00806
Analysis of Phylogenetic Variation of Stenotrophomonas maltophilia Reveals Human-Specific Branches
Abstract
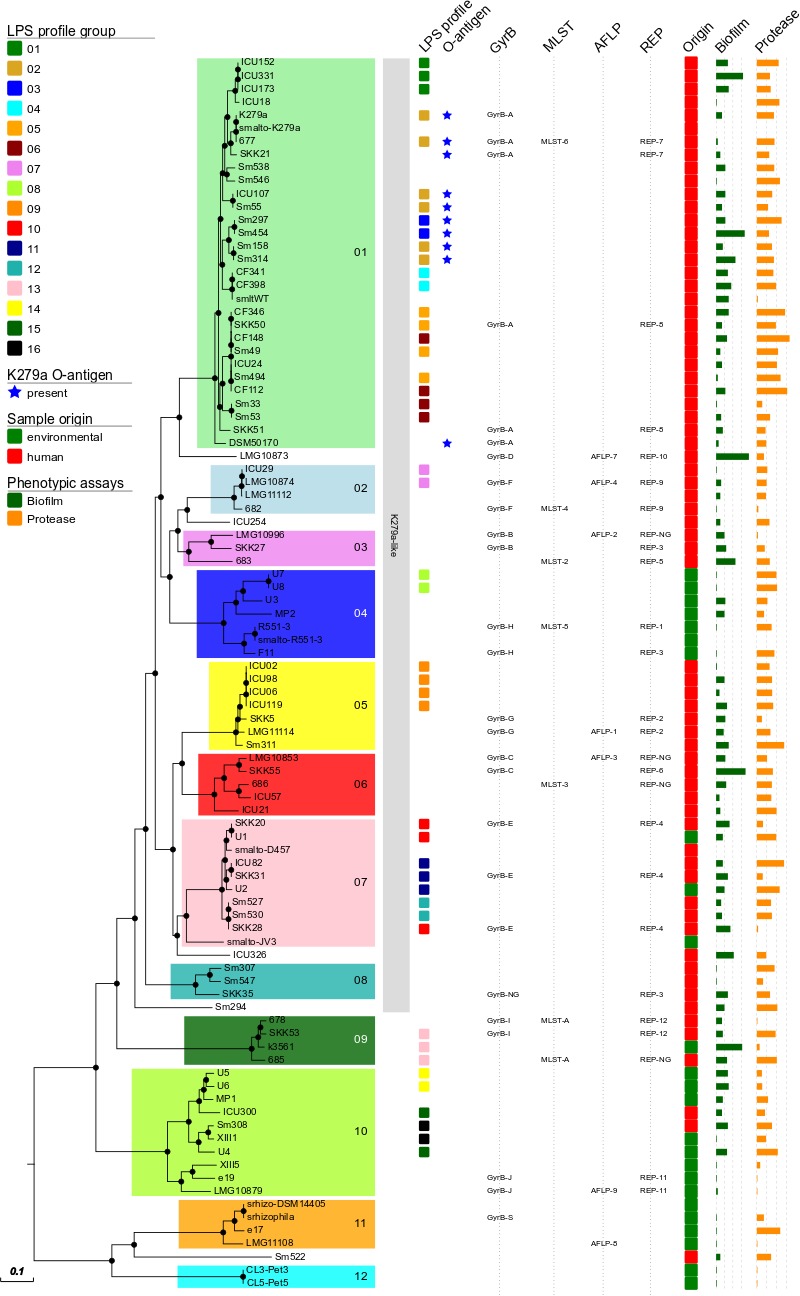
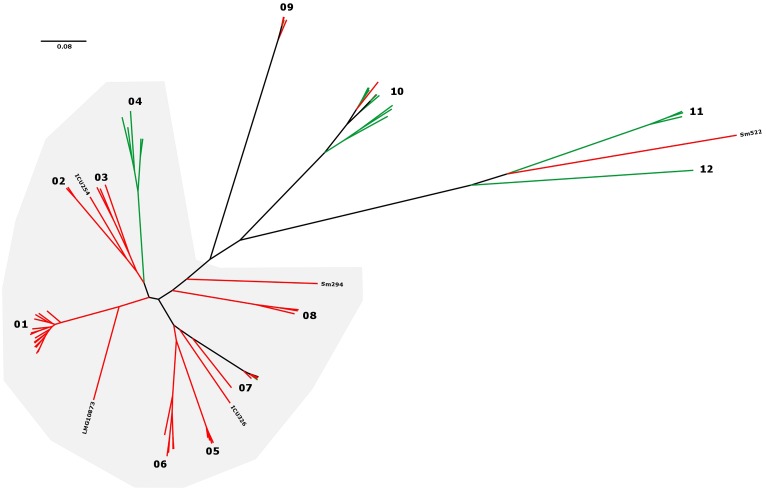
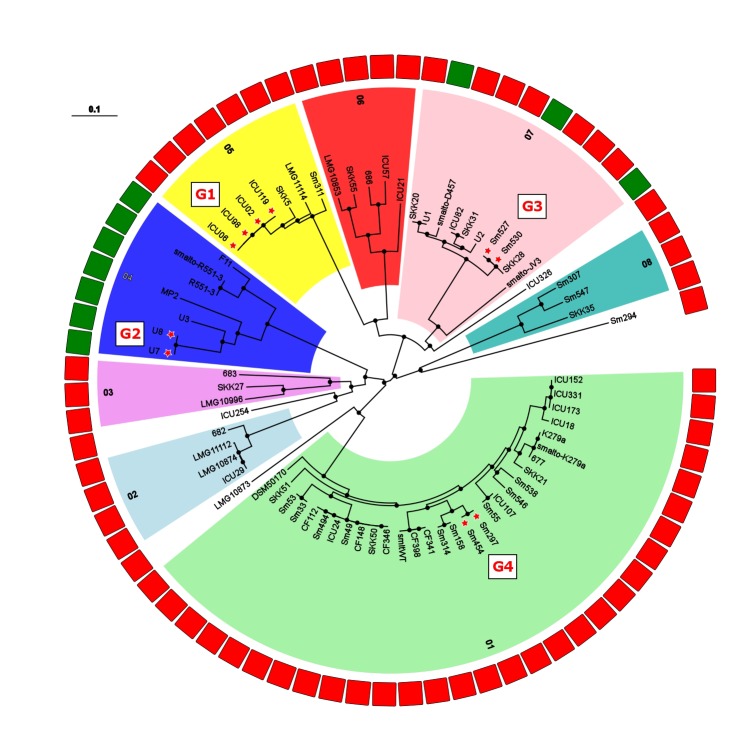
Stenotrophomonas maltophilia is a non-fermenting Gram-negative bacterium that is ubiquitous in the environment. In humans, this opportunistic multi-drug-resistant pathogen is responsible for a plethora of healthcare-associated infections. Here, we utilized a whole genome sequencing (WGS)-based phylogenomic core single nucleotide polymorphism (SNP) approach to characterize S. maltophilia subgroups, their potential association with human infection, and to detect any possible transmission events. In total, 89 isolates (67 clinical and 22 environmental) from Germany were sequenced. Fully finished genomes of five strains were included in the dataset for the core SNP phylogenomic analysis. WGS data were compared with conventional genotyping results as well as with underlying disease, biofilm formation, protease activity, lipopolysaccharide (LPS) SDS-PAGE profiles, and serological specificity of an antibody raised against the surface-exposed O-antigen of strain S. maltophilia K279a. The WGS-based phylogenies grouped the strains into 12 clades, out of which 6 contained exclusively human and 3 exclusively environmental isolates. Biofilm formation and proteolytic activity did correlate neither with the phylogenetic tree, nor with the origin of isolates. In contrast, the genomic classification correlated well with the reactivity of the strains against the K279a O-specific antibody, as well as in part with the LPS profiles. Three clusters of clinical strains had a maximum distance of 25 distinct SNP positions, pointing to possible transmission events or acquisition from the same source. In conclusion, these findings indicate the presence of specific subgroups of S. maltophilia strains adapted to the human host.
Keywords: Stenotrophomonas maltophilia; biofilm; cystic fibrosis; lipopolysaccharide; next generation sequencing; proteolytic activity; whole genome sequencing.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases