

Spatial mutation patterns as markers of early colorectal tumor cell mobility
- PMID: 29760052
- PMCID: PMC5984490
- DOI: 10.1073/pnas.1716552115
Spatial mutation patterns as markers of early colorectal tumor cell mobility
Abstract
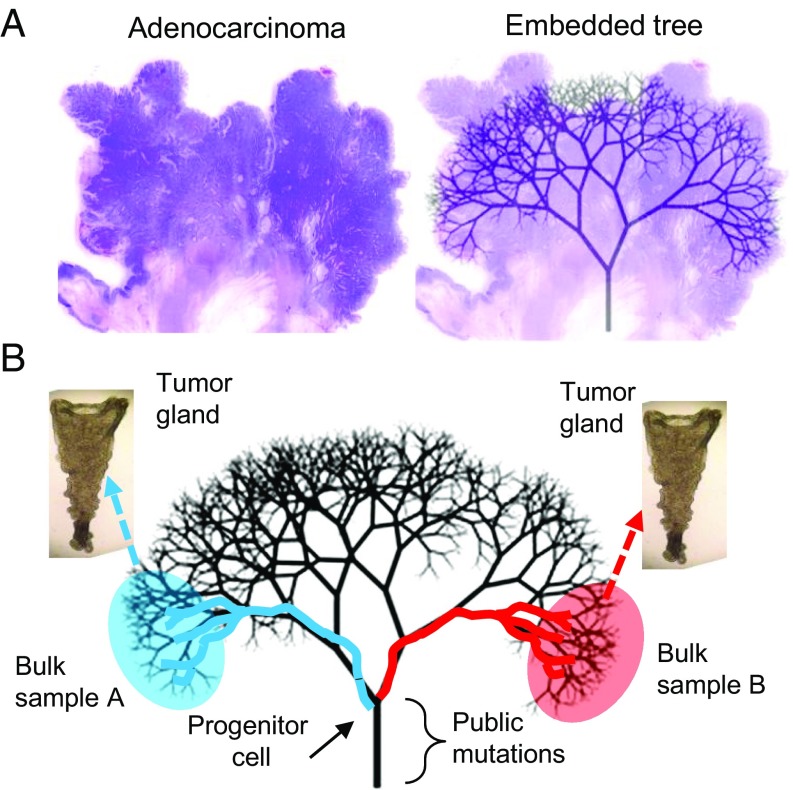
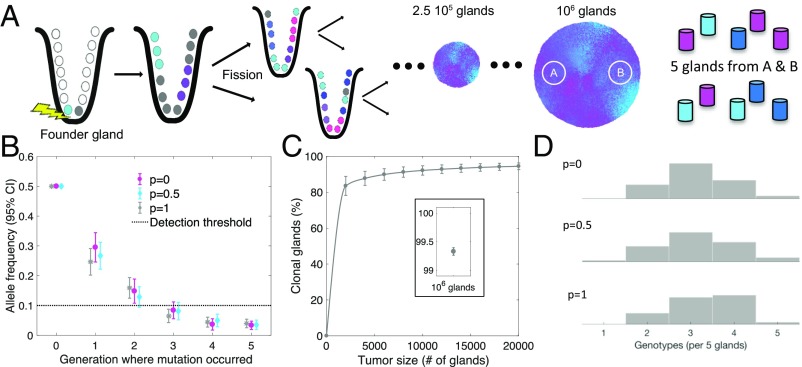
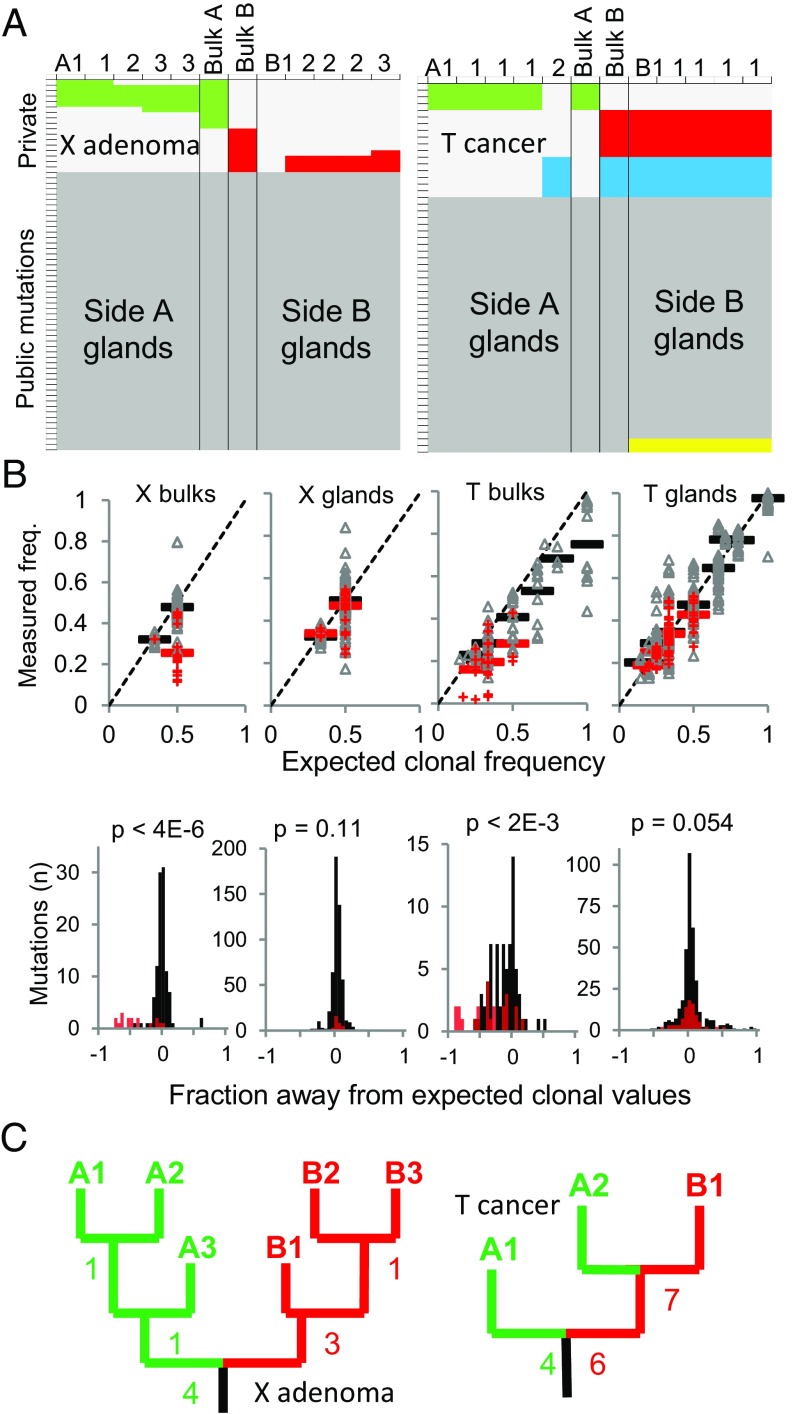
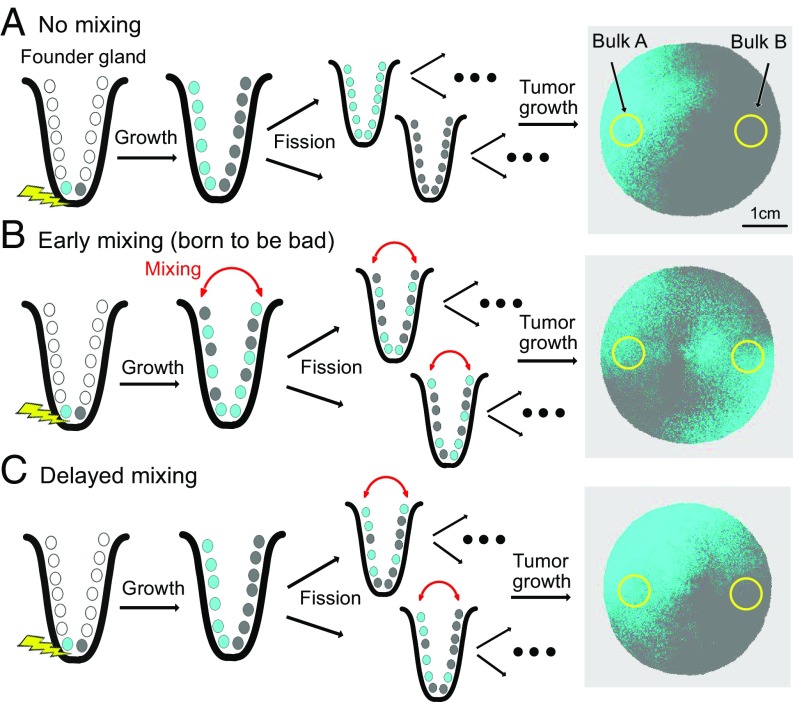
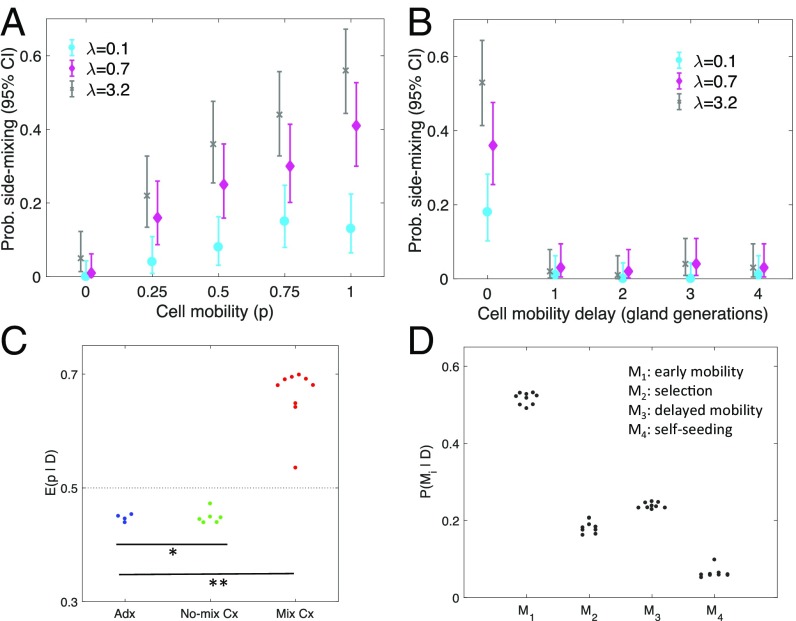
A growing body of evidence suggests that a subset of human cancers grows as single clonal expansions. In such a nearly neutral evolution scenario, it is possible to infer the early ancestral tree of a full-grown tumor. We hypothesized that early tree reconstruction can provide insights into the mobility phenotypes of tumor cells during their first few cell divisions. We explored this hypothesis by means of a computational multiscale model of tumor expansion incorporating the glandular structure of colorectal tumors. After calibrating the model to multiregional and single gland data from 19 human colorectal tumors using approximate Bayesian computation, we examined the role of early tumor cell mobility in shaping the private mutation patterns of the final tumor. The simulations showed that early cell mixing in the first tumor gland can result in side-variegated patterns where the same private mutations could be detected on opposite tumor sides. In contrast, absence of early mixing led to nonvariegated, sectional mutation patterns. These results suggest that the patterns of detectable private mutations in colorectal tumors may be a marker of early cell movement and hence the invasive and metastatic potential of the tumor at the start of the growth. In alignment with our hypothesis, we found evidence of early abnormal cell movement in 9 of 15 invasive colorectal carcinomas ("born to be bad"), but in none of 4 benign adenomas. If validated with a larger dataset, the private mutation patterns may be used for outcome prediction among screen-detected lesions with unknown invasive potential.
Keywords: cancer modeling; cellular mobility; intratumor heterogeneity; tumor evolution.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
