

Recent Advances in the Diagnosis and Pathogenesis of Neurofibromatosis Type 1 (NF1)-associated Peripheral Nervous System Neoplasms
- PMID: 29762158
- PMCID: PMC9216180
- DOI: 10.1097/PAP.0000000000000197
Recent Advances in the Diagnosis and Pathogenesis of Neurofibromatosis Type 1 (NF1)-associated Peripheral Nervous System Neoplasms
Abstract
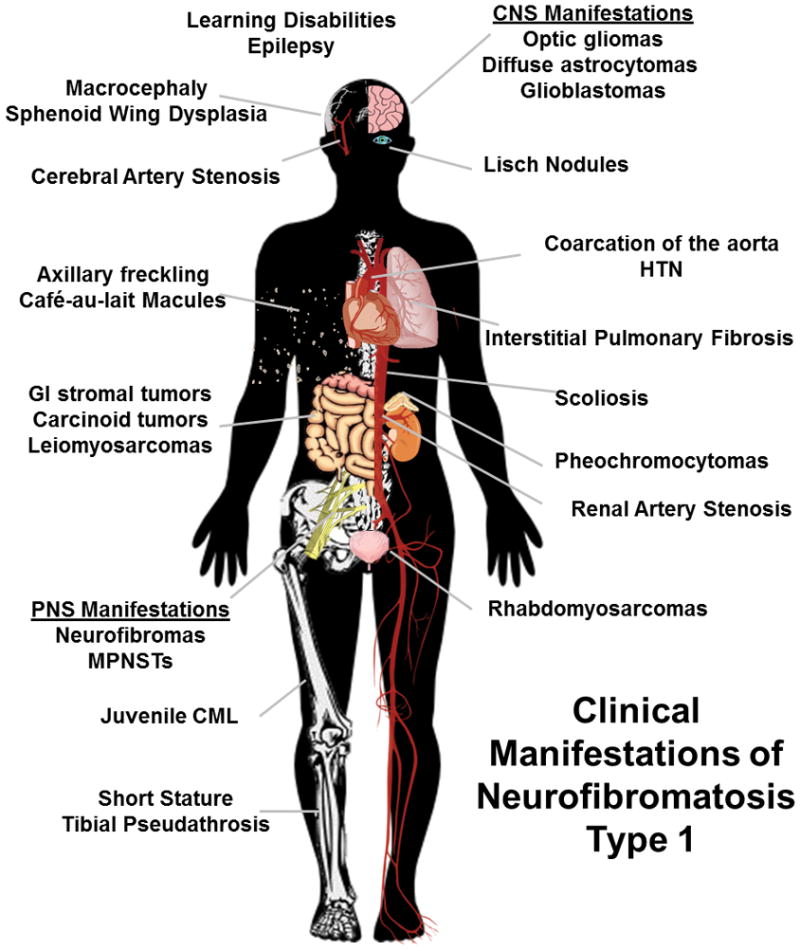
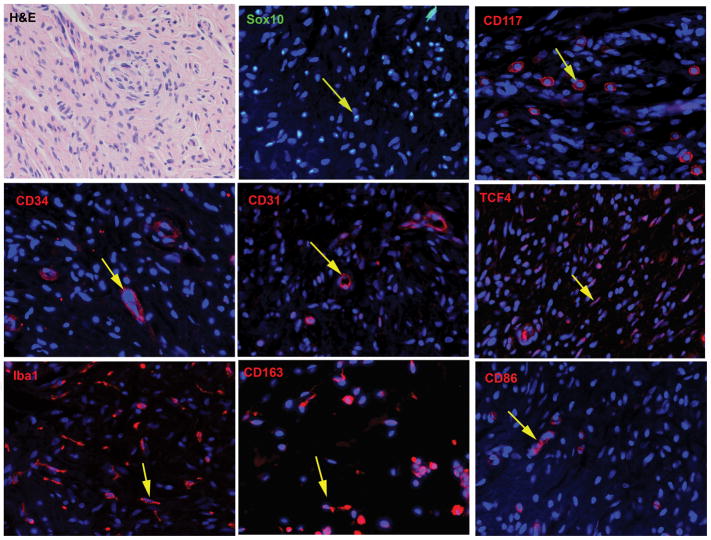
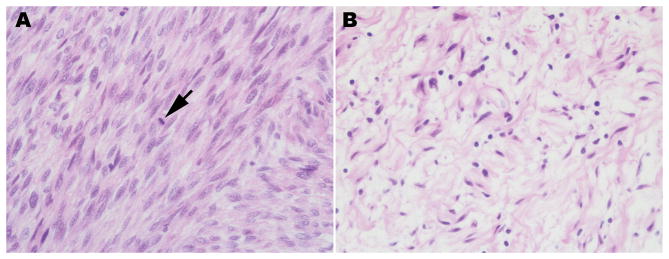
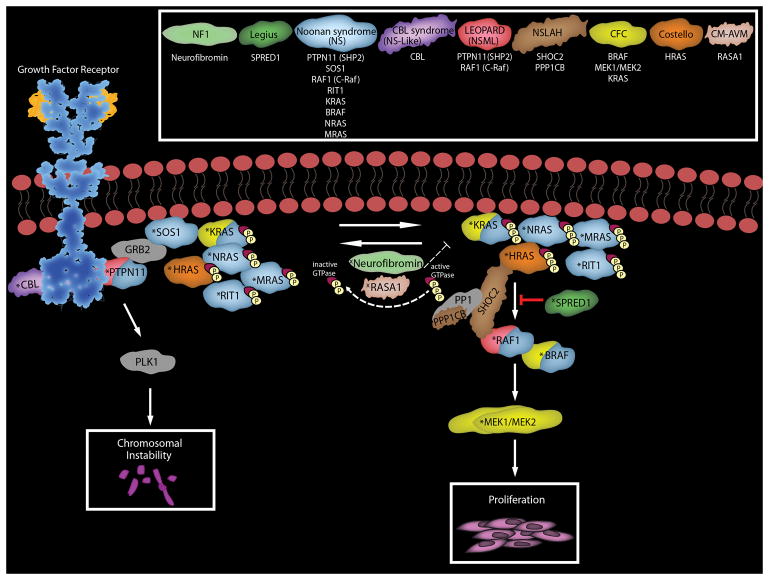
The diagnosis of a neurofibroma or a malignant peripheral nerve sheath tumor (MPNST) often raises the question of whether the patient has the genetic disorder neurofibromatosis type 1 (NF1) as well as how this will impact the patient's outcome, what their risk is for developing additional neoplasms and whether treatment options differ for NF1-associated and sporadic peripheral nerve sheath tumors. Establishing a diagnosis of NF1 is challenging as this disorder has numerous neoplastic and non-neoplastic manifestations which are variably present in individual patients. Further, other genetic diseases affecting the Ras signaling cascade (RASopathies) mimic many of the clinical features of NF1. Here, we review the clinical manifestations of NF1 and compare and contrast them with those of the RASopathies. We also consider current approaches to genetic testing for germline NF1 mutations. We then focus on NF1-associated neurofibromas, considering first the complicated clinical behavior and pathology of these neoplasms and then discussing our current understanding of the genomic abnormalities that drive their pathogenesis, including the mutations encountered in atypical neurofibromas. As several neurofibroma subtypes are capable of undergoing malignant transformation to become MPNSTs, we compare and contrast patient outcomes in sporadic, NF1-associated and radiation-induced MPNSTs, and review the challenging pathology of these lesions. The mutations involved in neurofibroma-MPNST progression, including the recent identification of mutations affecting epigenetic regulators, are then considered. Finally, we explore how our current understanding of neurofibroma and MPNST pathogenesis is informing the design of new therapies for these neoplasms.
Conflict of interest statement
Figures
References
-
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123:124–33. - PubMed
-
- Jadayel D, Fain P, Upadhyaya M, Ponder MA, Huson SM, Carey J, Fryer A, Mathew CG, Barker DF, Ponder BA. Paternal origin of new mutations in von Recklinghausen neurofibromatosis. Nature. 1990;343:558–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
