

Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells
- PMID: 29762492
- PMCID: PMC5977252
- DOI: 10.3390/v10050259
Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells
Abstract
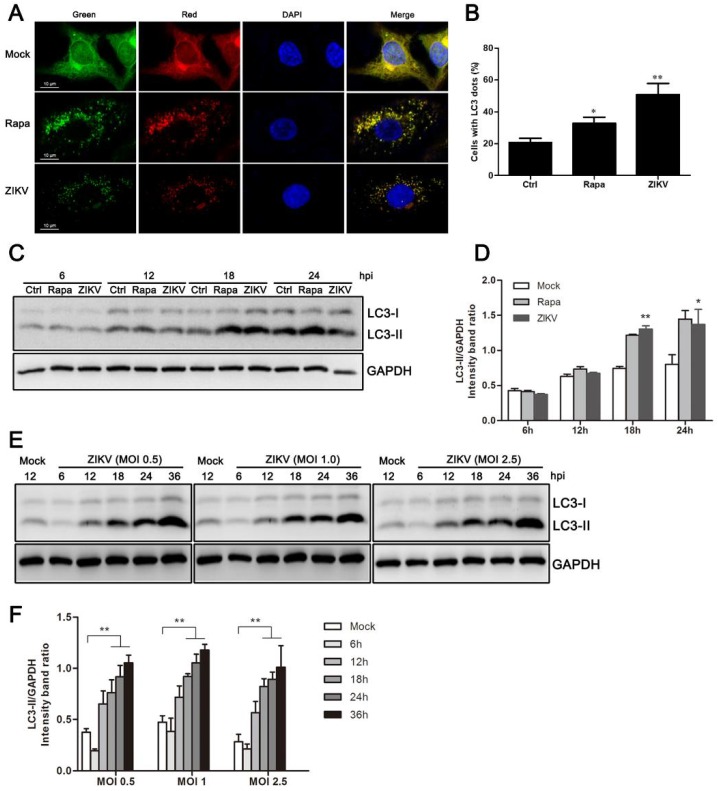
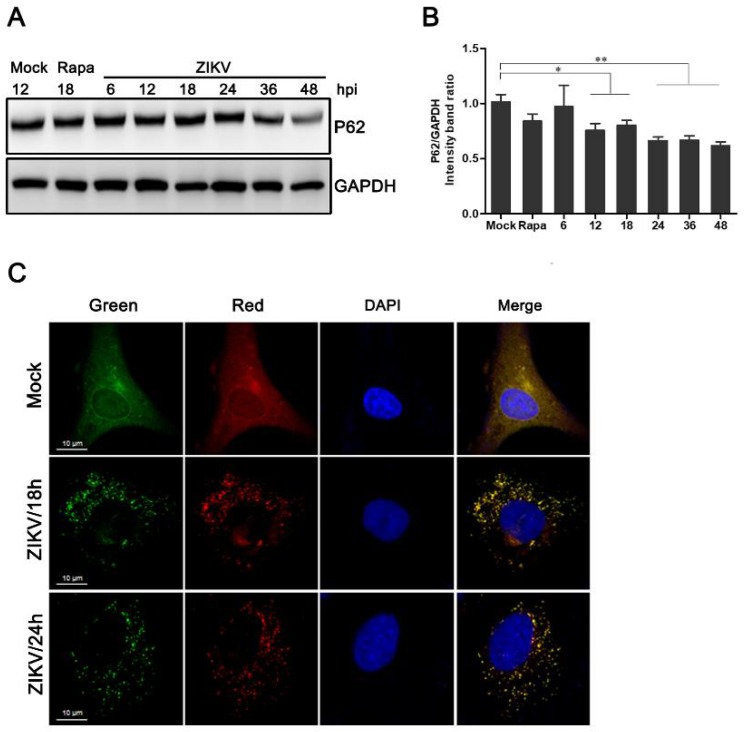
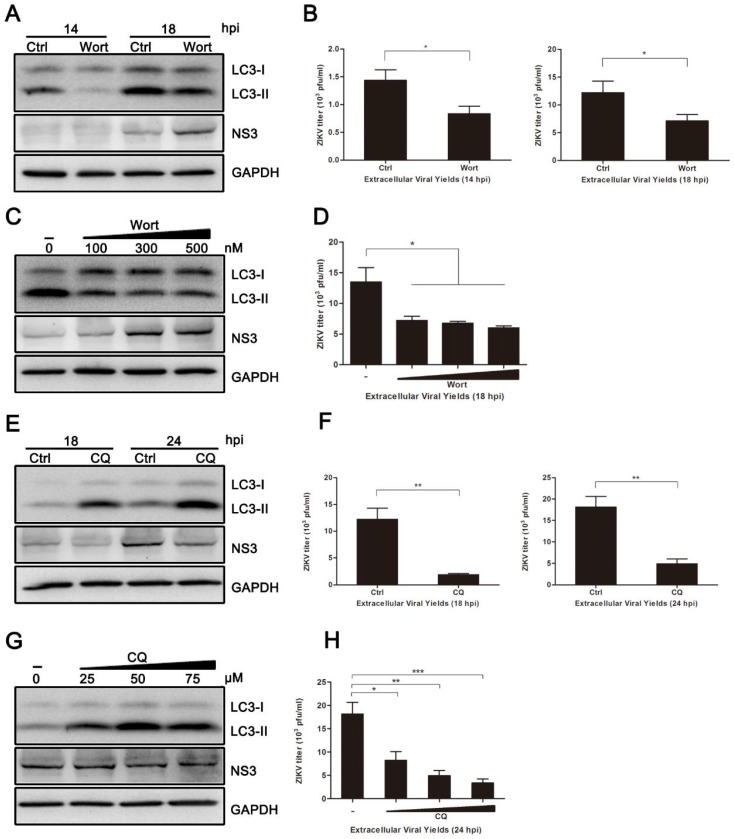
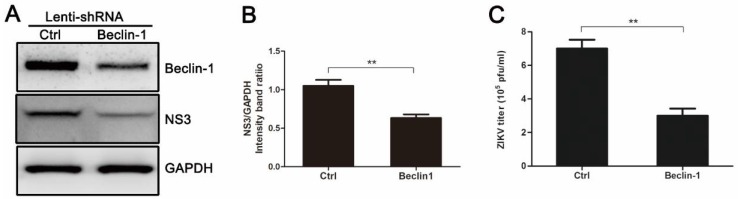
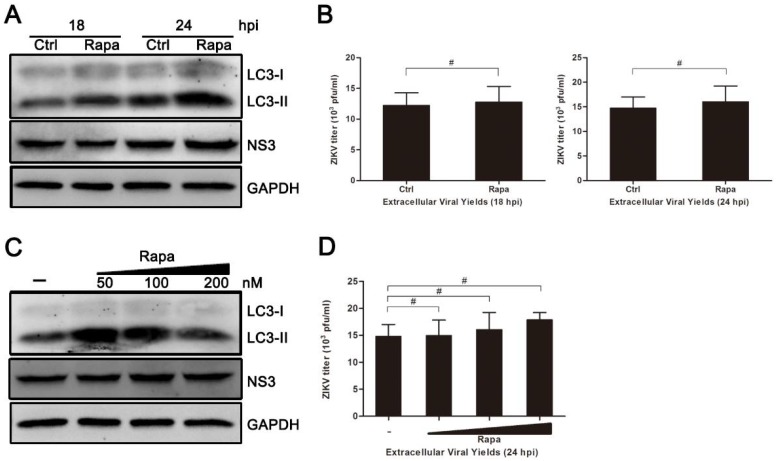
Autophagy is a common strategy for cell protection; however, some viruses can in turn adopt cellular autophagy to promote viral replication. Zika virus (ZIKV) is the pathogen that causes Zika viral disease, and it is a mosquito-borne virus. However, its pathogenesis, especially the interaction between ZIKV and target cells during the early stages of infection, is still unclear. In this study, we demonstrate that infecting human umbilical vein endothelial cells (HUVEC) with ZIKV triggers cellular autophagy. We observed both an increase in the conversion of LC3-I to LC3-II and increased accumulation of fluorescent cells with LC3 dots, which are considered to be the two key indicators of autophagy. The ratio of LC3-II/GAPDH in each group was significantly increased at different times after ZIKV infection at different MOIs, indicating that the production of lipidated LC3-II increased. Moreover, both the ratio of LC3-II/GAPDH and the expression of viral NS3 protein increased with increasing time of viral infection. The expression level of p62 decreased gradually from 12 h post-infection. Expression profile of double fluorescent protein labelling LC3 indicated that the autophagy induced by ZIKV infection was a complete process. We further investigated the role of autophagy in ZIKV replication. We demonstrated that either the treatment with inhibitors of autophagosomes formation or short hairpin RNA targeting the Beclin-1 gene, which is critical for the formation of autophagosomes, significantly reduced viral production. Taken together, our results indicate that ZIKV infection induces autophagy of HUVEC, and inhibition of ZIKV-induced autophagy restrains viral replication.
Keywords: Beclin-1; LC3; Zika virus; autophagy; viral replication; virus production.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Chouin-Carneiro T., Vega-Rua A., Vazeille M., Yebakima A., Girod R., Goindin D., Dupont-Rouzeyrol M., Lourenco-de-Oliveira R., Failloux A.B. Differential Susceptibilities of Aedes aegypti and Aedes albopictus from the Americas to Zika Virus. PLoS Negl. Trop. Dis. 2016;10:e0004543. doi: 10.1371/journal.pntd.0004543. - DOI - PMC - PubMed
-
- Calvet G., Aguiar R.S., Melo A.S., Sampaio S.A., de Filippis I., Fabri A., Araujo E.S., de Sequeira P.C., de Mendonca M.C., de Oliveira L., et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: A case study. Lancet Infect. Dis. 2016;16:653–660. doi: 10.1016/S1473-3099(16)00095-5. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
