

Optimized distributed systems achieve significant performance improvement on sorted merging of massive VCF files
- PMID: 29762754
- PMCID: PMC6007233
- DOI: 10.1093/gigascience/giy052
Optimized distributed systems achieve significant performance improvement on sorted merging of massive VCF files
Abstract
Background: Sorted merging of genomic data is a common data operation necessary in many sequencing-based studies. It involves sorting and merging genomic data from different subjects by their genomic locations. In particular, merging a large number of variant call format (VCF) files is frequently required in large-scale whole-genome sequencing or whole-exome sequencing projects. Traditional single-machine based methods become increasingly inefficient when processing large numbers of files due to the excessive computation time and Input/Output bottleneck. Distributed systems and more recent cloud-based systems offer an attractive solution. However, carefully designed and optimized workflow patterns and execution plans (schemas) are required to take full advantage of the increased computing power while overcoming bottlenecks to achieve high performance.
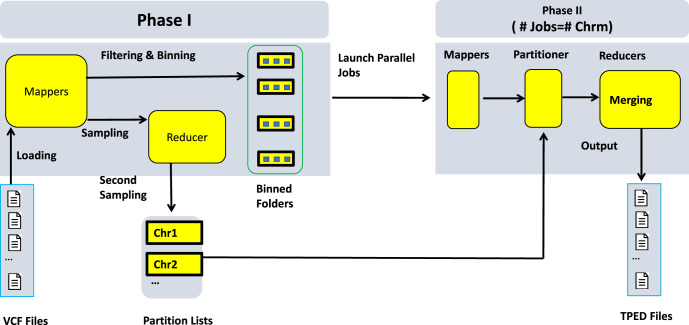
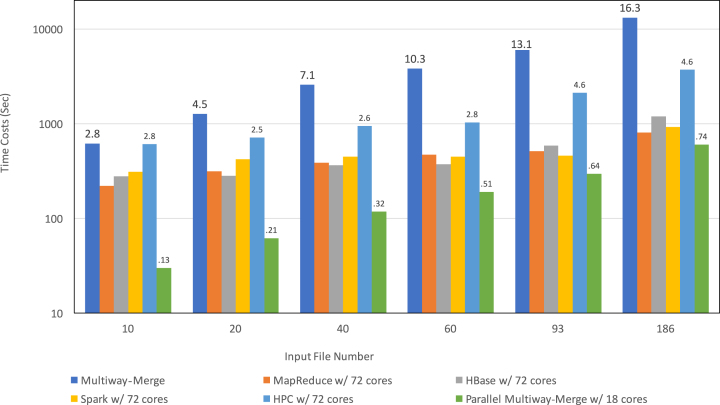
Findings: In this study, we custom-design optimized schemas for three Apache big data platforms, Hadoop (MapReduce), HBase, and Spark, to perform sorted merging of a large number of VCF files. These schemas all adopt the divide-and-conquer strategy to split the merging job into sequential phases/stages consisting of subtasks that are conquered in an ordered, parallel, and bottleneck-free way. In two illustrating examples, we test the performance of our schemas on merging multiple VCF files into either a single TPED or a single VCF file, which are benchmarked with the traditional single/parallel multiway-merge methods, message passing interface (MPI)-based high-performance computing (HPC) implementation, and the popular VCFTools.
Conclusions: Our experiments suggest all three schemas either deliver a significant improvement in efficiency or render much better strong and weak scalabilities over traditional methods. Our findings provide generalized scalable schemas for performing sorted merging on genetics and genomics data using these Apache distributed systems.
Figures








References
-
- Massie M, Nothaft F, Hartl C et al. . Adam: Genomics formats and processing patterns for cloud scale computing. University of California, Berkeley Technical Report, No UCB/EECS-2013 2013;207.
-
- Reyes-Ortiz JL, Oneto L, Anguita D. Big data analytics in the cloud: Spark on hadoop vs mpi/openmp on beowulf. Procedia Computer Science. 2015;53:121–30.
Publication types
MeSH terms
Grants and funding
- P01 NS097206/NS/NINDS NIH HHS/United States
- R01 HL128439/HL/NHLBI NIH HHS/United States
- U54 NS091859/NS/NINDS NIH HHS/United States
- R01 HL135156/HL/NHLBI NIH HHS/United States
- P60 MD006902/MD/NIMHD NIH HHS/United States
- R01 HL104608/HL/NHLBI NIH HHS/United States
- R01 MD010443/MD/NIMHD NIH HHS/United States
- R21 ES024844/ES/NIEHS NIH HHS/United States
- RL5 GM118984/GM/NIGMS NIH HHS/United States
- R01 HL117004/HL/NHLBI NIH HHS/United States
- R01 NS051630/NS/NINDS NIH HHS/United States
- U54 GM115428/GM/NIGMS NIH HHS/United States
- R01 ES015794/ES/NIEHS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
