

The Understanding and Management of Organism Toxicity in Septic Shock
- PMID: 29763894
- PMCID: PMC6785648
- DOI: 10.1159/000487818
The Understanding and Management of Organism Toxicity in Septic Shock
Abstract
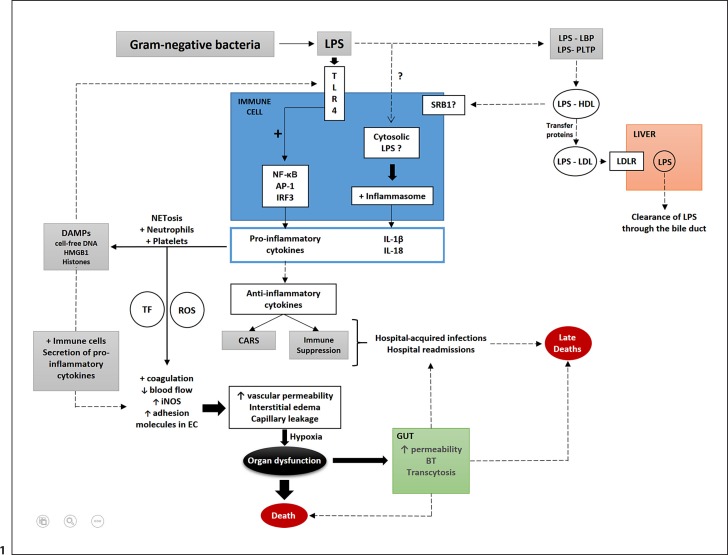
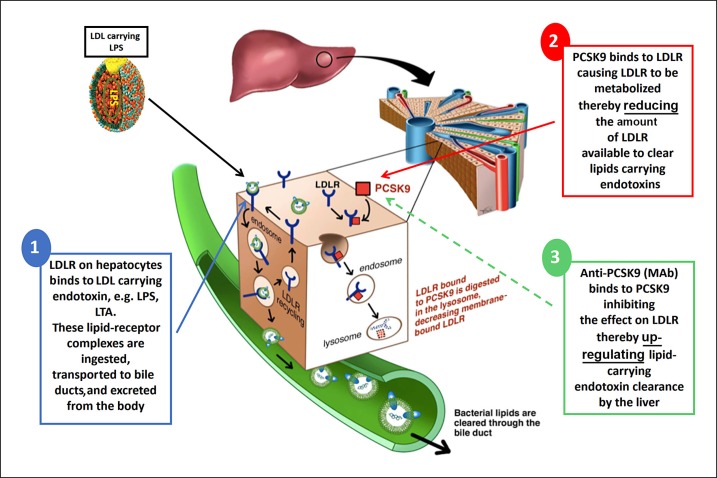
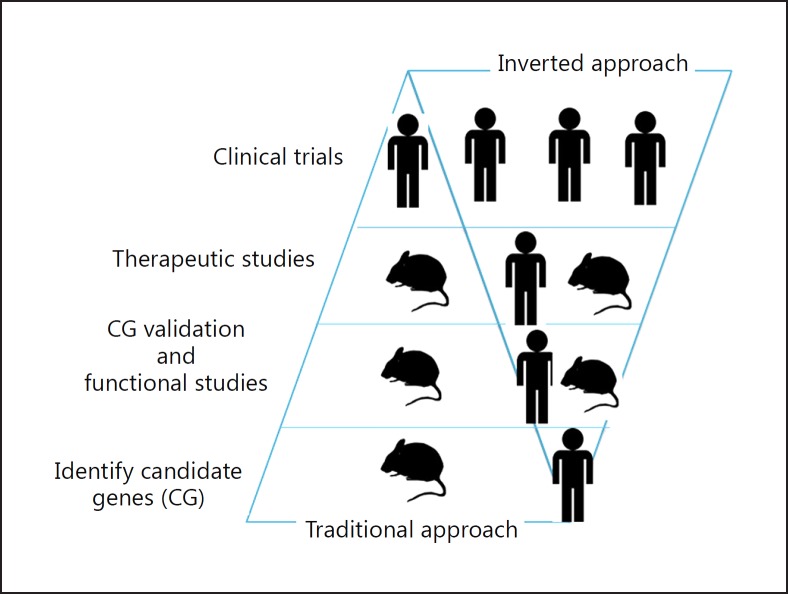
The toxicity caused by different organisms in septic shock is substantially complex and characterized by an intricate pathogenicity that involves several systems and pathways. Immune cells' pattern recognition receptors initiate the host response to pathogens after the recognition of pathogen-associated molecular patterns. In essence, the subsequent activation of downstream pathways may progress to infection resolution or to a dysregulated host response that represents the hallmark of organ injury in septic shock. Likewise, the management of organism toxicity in septic shock is complicated and comprises a multiplicity of suitable targets. In this review, the classic immune responses to pathogens are discussed as well as other factors that are relevant in the pathogenicity of septic shock, including sepsis-induced immune suppression, inflammasome activation, intestinal permeability, and the role of lipids and proprotein convertase subtilisin/kexin type 9. Current therapies aiming to eliminate the organisms causing septic shock, recent and ongoing trials in septic shock treatment, and potential new therapeutic strategies are also explored.
Keywords: Mediators; PCSK9; Sepsis; Septic shock; Therapies.
© 2018 S. Karger AG, Basel.
Figures
References
-
- Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. - PubMed
-
- Creagh EM, O'Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27:352–357. - PubMed
-
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. - PubMed
-
- Adib-Conquy M, Cavaillon JM. Compensatory anti-inflammatory response syndrome. Thromb Haemost. 2009;101:36–47. - PubMed
-
- Jin HS, Park JK, Jo EK. Toll-like receptors and NOD-like receptors in innate immune defense during pathogenic infection. J Bacteriol Virol. 2014;44:215–225.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
