

COL4A5 and LAMA5 variants co-inherited in familial hematuria: digenic inheritance or genetic modifier effect?
- PMID: 29764427
- PMCID: PMC5954460
- DOI: 10.1186/s12882-018-0906-5
COL4A5 and LAMA5 variants co-inherited in familial hematuria: digenic inheritance or genetic modifier effect?
Abstract
Background: About 40-50% of patients with familial microscopic hematuria (FMH) caused by thin basement membrane nephropathy (TBMN) inherit heterozygous mutations in collagen IV genes (COL4A3, COL4A4). On long follow-up, the full phenotypic spectrum of these patients varies a lot, ranging from isolated MH or MH plus low-grade proteinuria to chronic renal failure of variable degree, including end-stage renal disease (ESRD).
Methods: Here, we performed Whole Exome Sequencing (WES) in patients of six families, presenting with autosomal dominant FMH, with or without progression to proteinuria and loss of renal function, all previously found negative for severe collagen IV mutations. Hierarchical filtering of the WES data was performed, followed by mutation prediction analysis, Sanger sequencing and genetic segregation analysis.
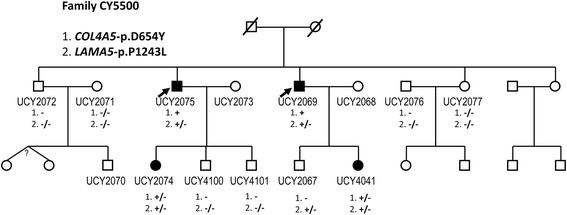
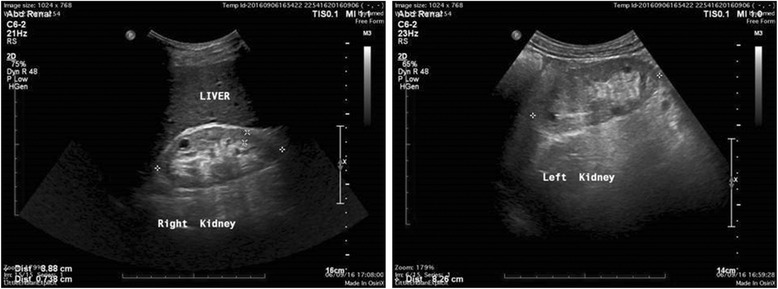
Results: In one family with four patients, we found evidence for the contribution of two co-inherited variants in two crucial genes expressed in the glomerular basement membrane (GBM); LAMA5-p.Pro1243Leu and COL4A5-p.Asp654Tyr. Mutations in COL4A5 cause classical X-linked Alport Syndrome, while rare mutations in the LAMA5 have been reported in patients with focal segmental glomerulosclerosis. The phenotypic spectrum of the patients includes hematuria, proteinuria, focal segmental glomerulosclerosis, loss of kidney function and renal cortical cysts.
Conclusions: A modifier role of LAMA5 on the background of a hypomorphic Alport syndrome causing mutation is a possible explanation of our findings. Digenic inheritance is another scenario, following the concept that mutations at both loci more accurately explain the spectrum of symptoms, but further investigation is needed under this concept. This is the third report linking a LAMA5 variant with human renal disease and expanding the spectrum of genes involved in glomerular pathologies accompanied by familial hematurias. The cystic phenotype overlaps with that of a mouse model, which carried a Lama5 hypomorphic mutation that caused severely reduced Lama5 protein levels and produced kidney cysts.
Keywords: Alport syndrome; Collagen IV; Digenic inheritance; FSGS; Familial hematuria; Kidney disease; Laminin alpha 5; Metalloproteinase; Modifier gene; Renal cysts; Synaptopodin; Thin Basement Membrane Nephropathy (TBMN).
Conflict of interest statement
Ethics approval and consent to participate
The study was approved by the Cyprus National Bioethics Committee and all participants gave a signed consent.
Consent for publication
All patients have provided written consent for their data to be used in a research publication and that this has also been verified and approved by the Cyprus National Bioethics Committee.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Haas M. Alport syndrome and thin glomerular basement membrane nephropathy: a practical approach to diagnosis. Arch Pathol Lab Med. 2009;133(2):224–232. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
