

Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes
- PMID: 29765031
- PMCID: PMC5953939
- DOI: 10.1038/s41467-018-04310-9
Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes
Abstract
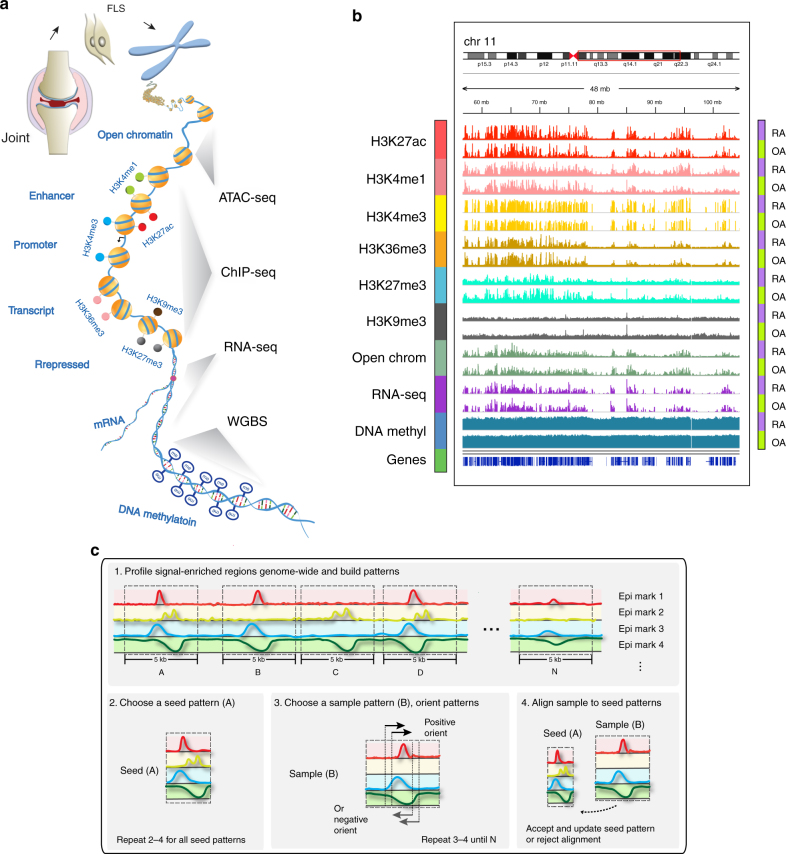
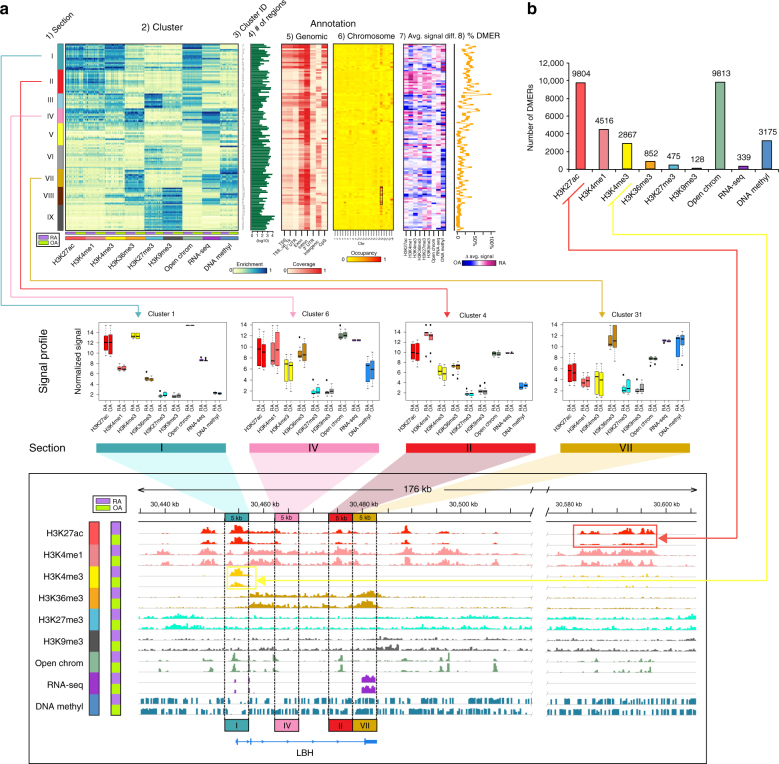
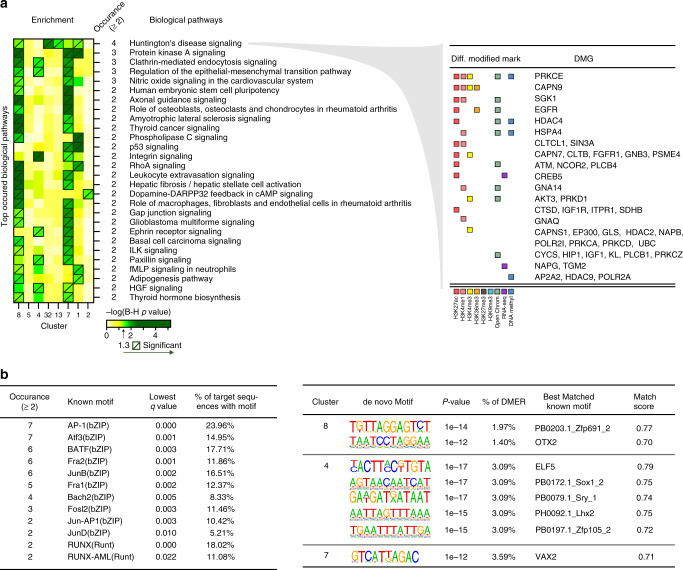
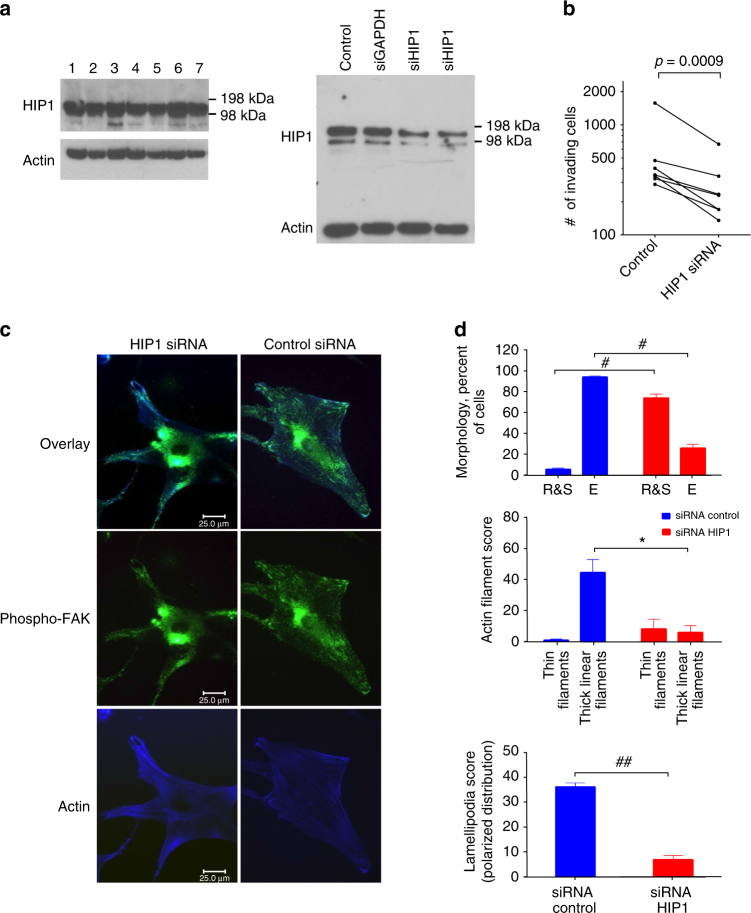
Epigenetics contributes to the pathogenesis of immune-mediated diseases like rheumatoid arthritis (RA). Here we show the first comprehensive epigenomic characterization of RA fibroblast-like synoviocytes (FLS), including histone modifications (H3K27ac, H3K4me1, H3K4me3, H3K36me3, H3K27me3, and H3K9me3), open chromatin, RNA expression and whole-genome DNA methylation. To address complex multidimensional relationship and reveal epigenetic regulation of RA, we perform integrative analyses using a novel unbiased method to identify genomic regions with similar profiles. Epigenomically similar regions exist in RA cells and are associated with active enhancers and promoters and specific transcription factor binding motifs. Differentially marked genes are enriched for immunological and unexpected pathways, with "Huntington's Disease Signaling" identified as particularly prominent. We validate the relevance of this pathway to RA by showing that Huntingtin-interacting protein-1 regulates FLS invasion into matrix. This work establishes a high-resolution epigenomic landscape of RA and demonstrates the potential for integrative analyses to identify unanticipated therapeutic targets.
Conflict of interest statement
E.P., V.K., D.P., J.W.W., Y.B., S.N., and K.E.B. are employees of Janssen Pharmaceuticals. All the remaining authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
