

Sodium Tanshinone IIA Sulfonate Decreases Cigarette Smoke-Induced Inflammation and Oxidative Stress via Blocking the Activation of MAPK/HIF-1α Signaling Pathway
- PMID: 29765317
- PMCID: PMC5938387
- DOI: 10.3389/fphar.2018.00263
Sodium Tanshinone IIA Sulfonate Decreases Cigarette Smoke-Induced Inflammation and Oxidative Stress via Blocking the Activation of MAPK/HIF-1α Signaling Pathway
Abstract
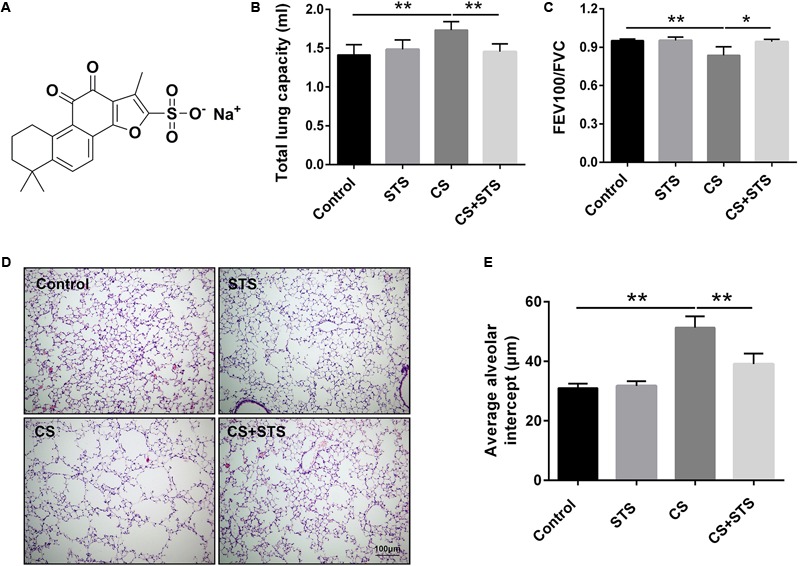
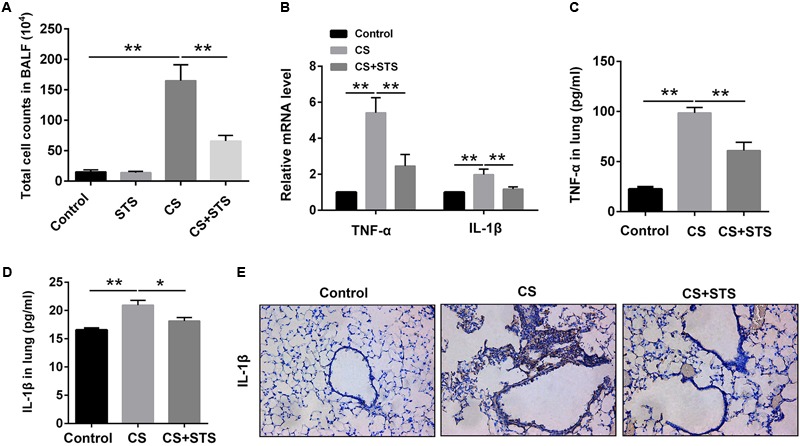
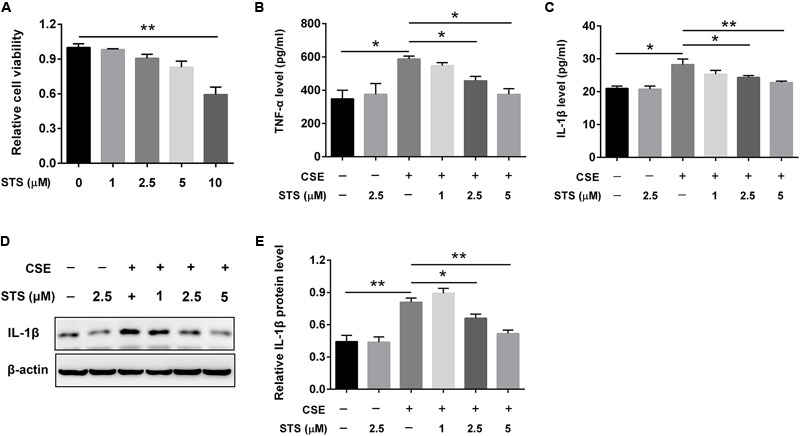
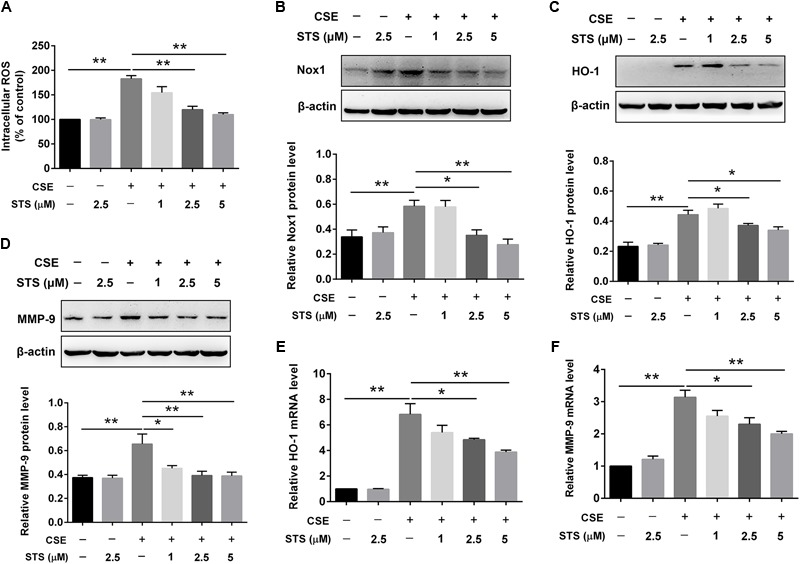
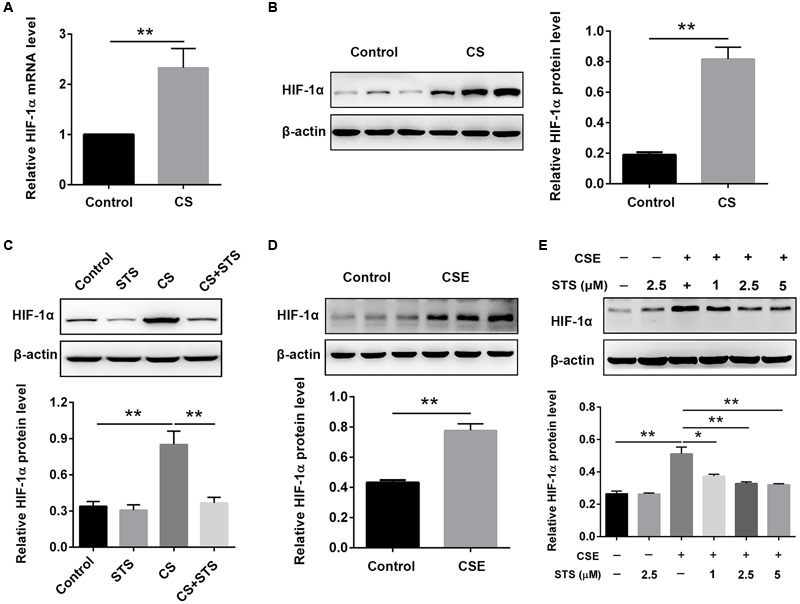
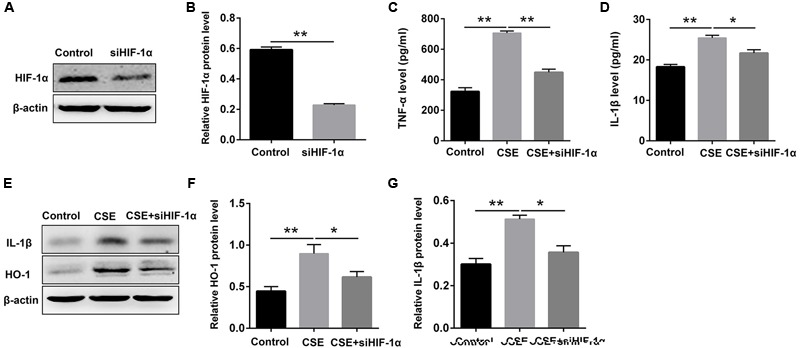
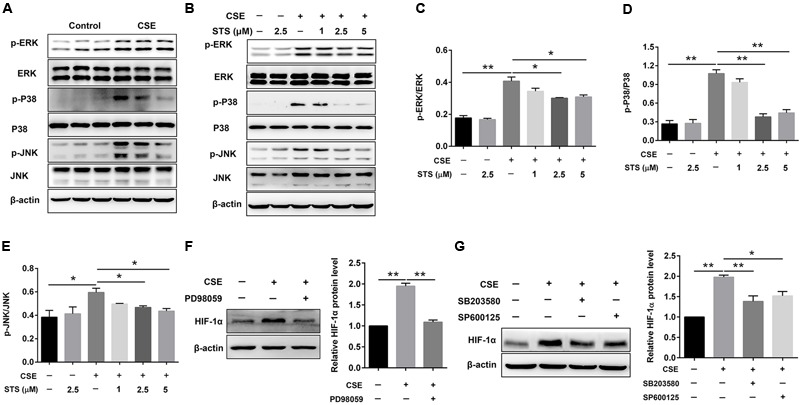
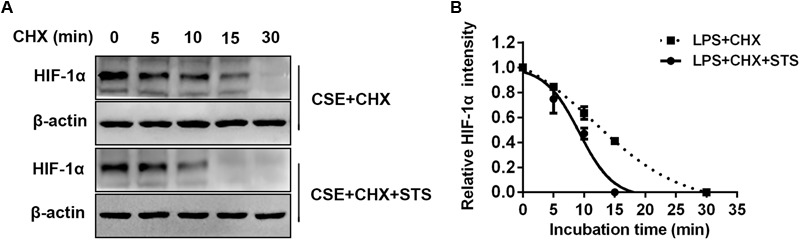
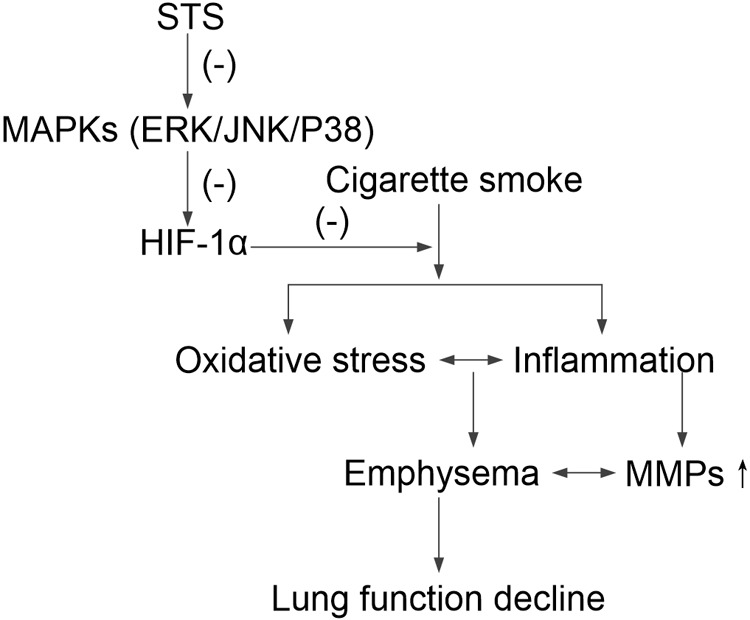
Aberrant activation of hypoxia-inducible factor (HIF)-1α is frequently encountered and promotes oxidative stress and inflammation in chronic obstructive pulmonary disease (COPD). The present study investigated whether sodium tanshinone IIA sulfonate (STS), a water-soluble derivative of tanshinone IIA, can mediate its effect through inhibiting HIF-1α-induced oxidative stress and inflammation in cigarette smoke (CS)-induced COPD in mice. Here, we found that STS improved pulmonary function, ameliorated emphysema and decreased the infiltration of inflammatory cells in the lungs of CS-exposed mice. STS reduced CS- and cigarette smoke extract (CSE)-induced upregulation of tumor necrosis factor (TNF)-α and interleukin (IL)-1β in the lungs and macrophages. STS also inhibited CSE-induced reactive oxygen species (ROS) production, as well as the upregulation of heme oxygenase (HO)-1, NOX1 and matrix metalloproteinase (MMP)-9 in macrophages. In addition, STS suppressed HIF-1α expression in vivo and in vitro, and pretreatment with HIF-1α siRNA reduced CSE-induced elevation of TNF-α, IL-1β, and HO-1 content in the macrophages. Moreover, we found that STS inhibited CSE-induced the phosphorylation of ERK, p38 MAPK and JNK in macrophages, and inhibition of these signaling molecules significantly repressed CSE-induced HIF-1α expression. It indicated that STS inhibits CSE-induced HIF-1α expression likely by blocking MAPK signaling. Furthermore, STS also promoted HIF-1α protein degradation in CSE-stimulated macrophages. Taken together, these results suggest that STS prevents COPD development possibly through the inhibition of HIF-1α signaling, and may be a novel strategy for the treatment of COPD.
Keywords: COPD; cigarette smoke; hypoxia-inducible factor-1α; inflammation; oxidative stress; sodium tanshinone IIA sulfonate.
Figures
References
-
- Barnes P. J., Shapiro S. D., Pauwels R. A. (2003). Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur. Respir. J. 22 672–688. - PubMed
-
- Bewley M. A., Belchamber K. B., Chana K. K., Budd R. C., Donaldson G., Wedzicha J. A., et al. (2016). Differential effects of p38, MAPK, PI3K or Rho Kinase inhibitors on bacterial phagocytosis and efferocytosis by macrophages in COPD. PLoS One 11:e0163139. 10.1371/journal.pone.0163139 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous