

Evolutionary Analysis Provides Insight Into the Origin and Adaptation of HCV
- PMID: 29765366
- PMCID: PMC5938362
- DOI: 10.3389/fmicb.2018.00854
Evolutionary Analysis Provides Insight Into the Origin and Adaptation of HCV
Abstract
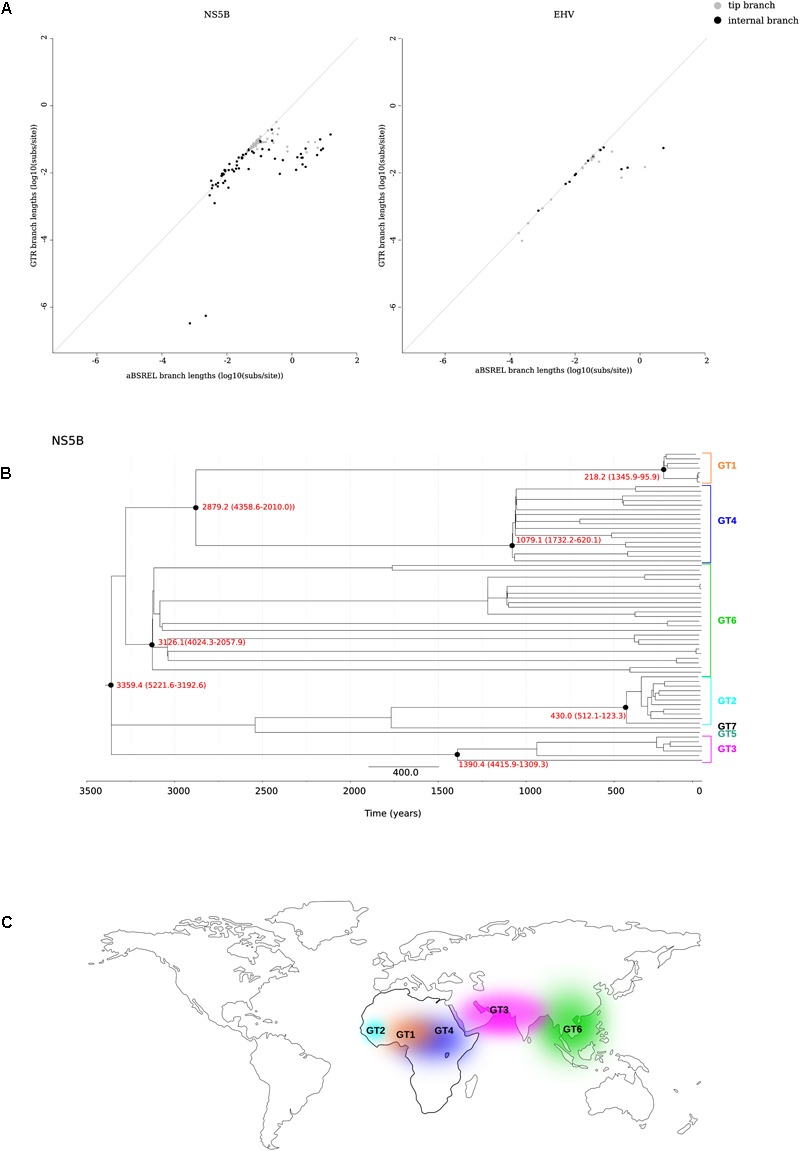
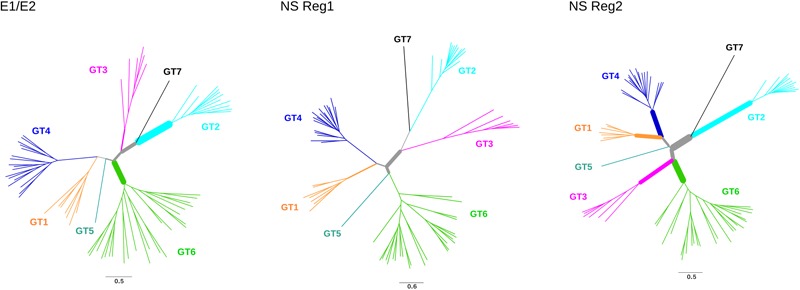
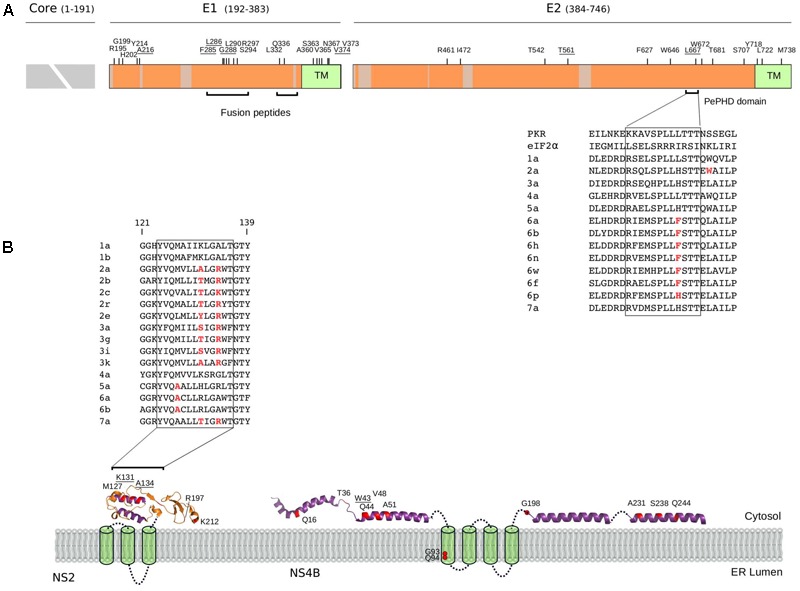
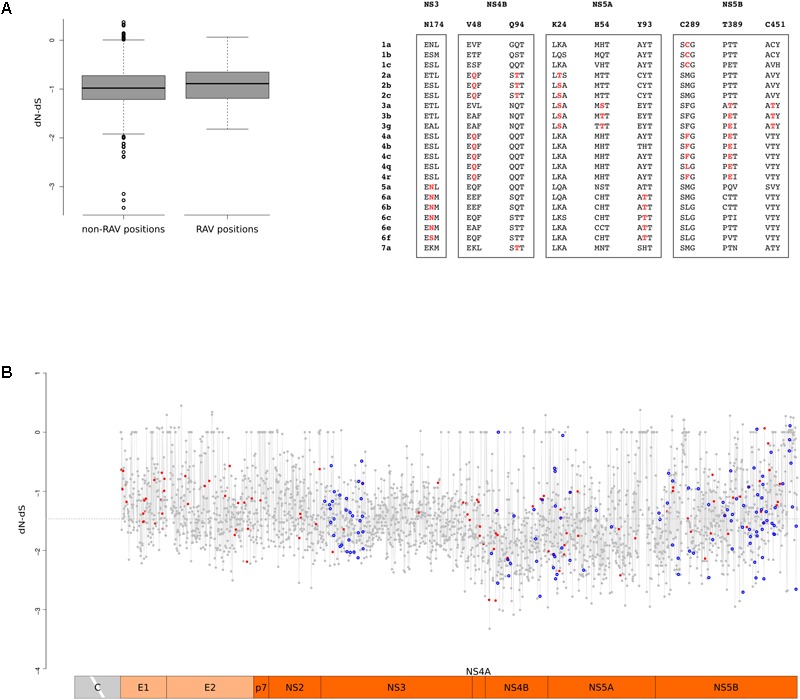
Hepatitis C virus (HCV) belongs to the Hepacivirus genus and is genetically heterogeneous, with seven major genotypes further divided into several recognized subtypes. HCV origin was previously dated in a range between ∼200 and 1000 years ago. Hepaciviruses have been identified in several domestic and wild mammals, the largest viral diversity being observed in bats and rodents. The closest relatives of HCV were found in horses/donkeys (equine hepaciviruses, EHV). However, the origin of HCV as a human pathogen is still an unsolved puzzle. Using a selection-informed evolutionary model, we show that the common ancestor of extant HCV genotypes existed at least 3000 years ago (CI: 3192-5221 years ago), with the oldest genotypes being endemic to Asia. EHV originated around 1100 CE (CI: 291-1640 CE). These time estimates exclude that EHV transmission was mainly sustained by widespread veterinary practices and suggest that HCV originated from a single zoonotic event with subsequent diversification in human populations. We also describe a number of biologically important sites in the major HCV genotypes that have been positively selected and indicate that drug resistance-associated variants are significantly enriched at positively selected sites. HCV exploits several cell-surface molecules for cell entry, but only two of these (CD81 and OCLN) determine the species-specificity of infection. Herein evolutionary analyses do not support a long-standing association between primates and hepaciviruses, and signals of positive selection at CD81 were only observed in Chiroptera. No evidence of selection was detected for OCLN in any mammalian order. These results shed light on the origin of HCV and provide a catalog of candidate genetic modulators of HCV phenotypic diversity.
Keywords: CD81; equine hepacivirus; hepatitis C virus; molecular dating; positive selection; resistance-associated amino acid variants; tMRCA.
Figures


References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous