

Core Clinical Phenotypes in Myotonic Dystrophies
- PMID: 29770119
- PMCID: PMC5941986
- DOI: 10.3389/fneur.2018.00303
Core Clinical Phenotypes in Myotonic Dystrophies
Abstract
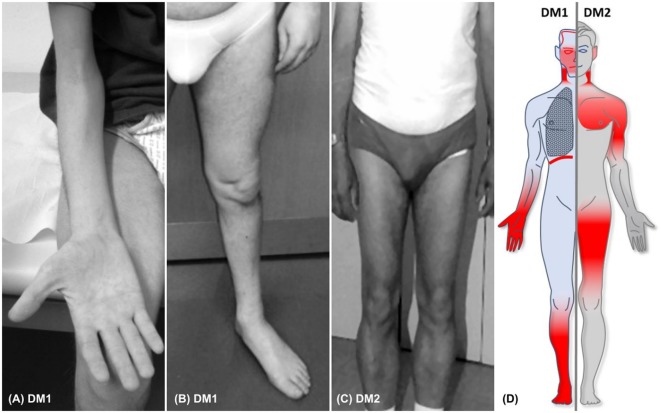
Myotonic dystrophy type 1 (DM1) and type 2 (DM2) represent the most frequent multisystemic muscular dystrophies in adulthood. They are progressive, autosomal dominant diseases caused by an abnormal expansion of an unstable nucleotide repeat located in the non-coding region of their respective genes DMPK for DM1 and CNBP in DM2. Clinically, these multisystemic disorders are characterized by a high variability of muscular and extramuscular symptoms, often causing a delay in diagnosis. For both subtypes, many symptoms overlap, but some differences allow their clinical distinction. This article highlights the clinical core features of myotonic dystrophies, thus facilitating their early recognition and diagnosis. Particular attention will be given to signs and symptoms of muscular involvement, to issues related to respiratory impairment, and to the multiorgan involvement. This article is part of a Special Issue entitled "Beyond Borders: Myotonic Dystrophies-A European Perception."
Keywords: DM1; DM2; myotonia; myotonic dystrophies; phenotypes; repeat expansion diseases; sleep disorders.
Figures

References
-
- Udd B, Meola G, Krahe R, Wansink DG, Bassez G, Kress W, et al. Myotonic dystrophy type 2 (DM2) and related disorders report of the 180th ENMC workshop including guidelines on diagnostics and management 3-5 December 2010, Naarden, The Netherlands. Neuromuscul Disord (2011) 21(6):443–50. 10.1016/j.nmd.2011.03.013 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous