

Targeting Oxidatively Induced DNA Damage Response in Cancer: Opportunities for Novel Cancer Therapies
- PMID: 29770165
- PMCID: PMC5892224
- DOI: 10.1155/2018/2389523
Targeting Oxidatively Induced DNA Damage Response in Cancer: Opportunities for Novel Cancer Therapies
Abstract
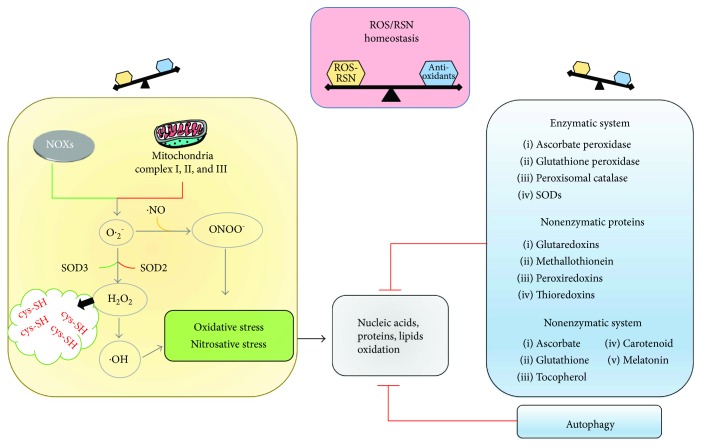
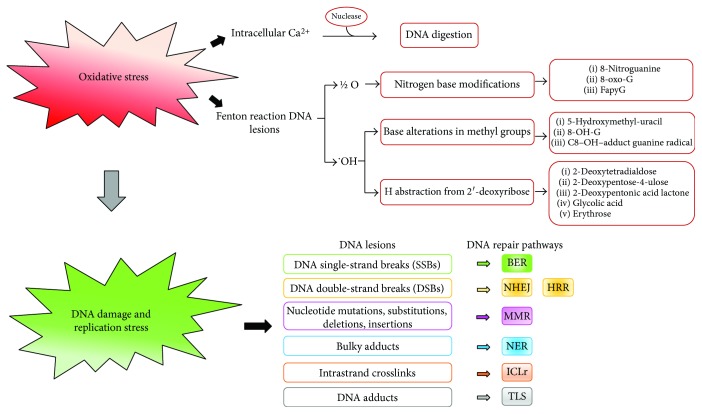
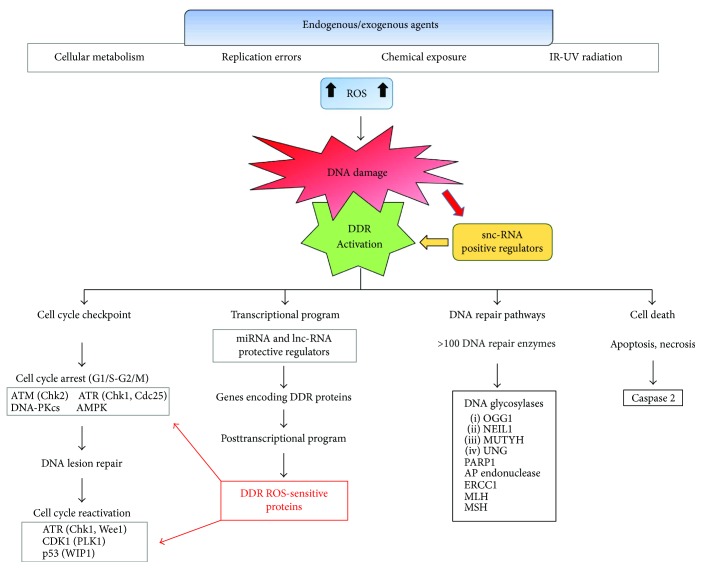
Cancer is a death cause in economically developed countries that results growing also in developing countries. Improved outcome through targeted interventions faces the scarce selectivity of the therapies and the development of resistance to them that compromise the therapeutic effects. Genomic instability is a typical cancer hallmark due to DNA damage by genetic mutations, reactive oxygen and nitrogen species, ionizing radiation, and chemotherapeutic agents. DNA lesions can induce and/or support various diseases, including cancer. The DNA damage response (DDR) is a crucial signaling-transduction network that promotes cell cycle arrest or cell death to repair DNA lesions. DDR dysregulation favors tumor growth as downregulated or defective DDR generates genomic instability, while upregulated DDR may confer treatment resistance. Redox homeostasis deeply and capillary affects DDR as ROS activate/inhibit proteins and enzymes integral to DDR both in healthy and cancer cells, although by different routes. DDR regulation through modulating ROS homeostasis is under investigation as anticancer opportunity, also in combination with other treatments since ROS affect DDR differently in the patients during cancer development and treatment. Here, we highlight ROS-sensitive proteins whose regulation in oxidatively induced DDR might allow for selective strategies against cancer that are better tailored to the patients.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
