

The Making of Leukemia
- PMID: 29772764
- PMCID: PMC5983781
- DOI: 10.3390/ijms19051494
The Making of Leukemia
Abstract
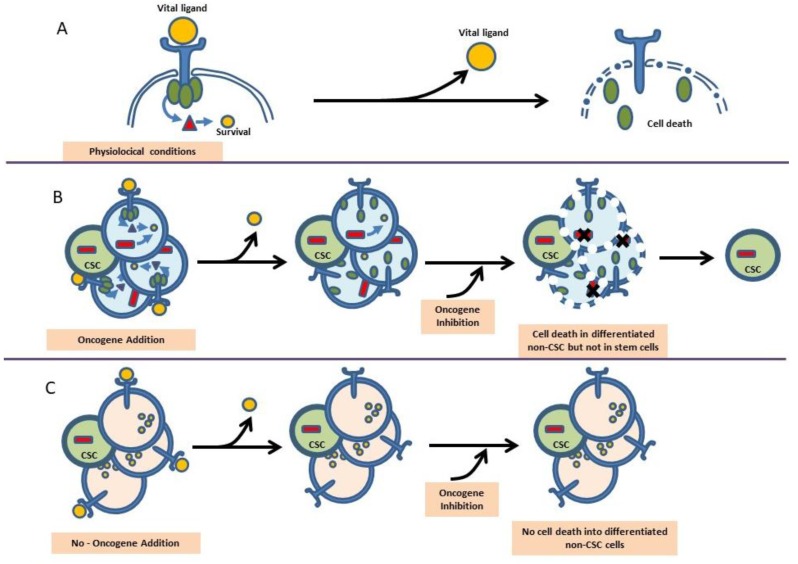
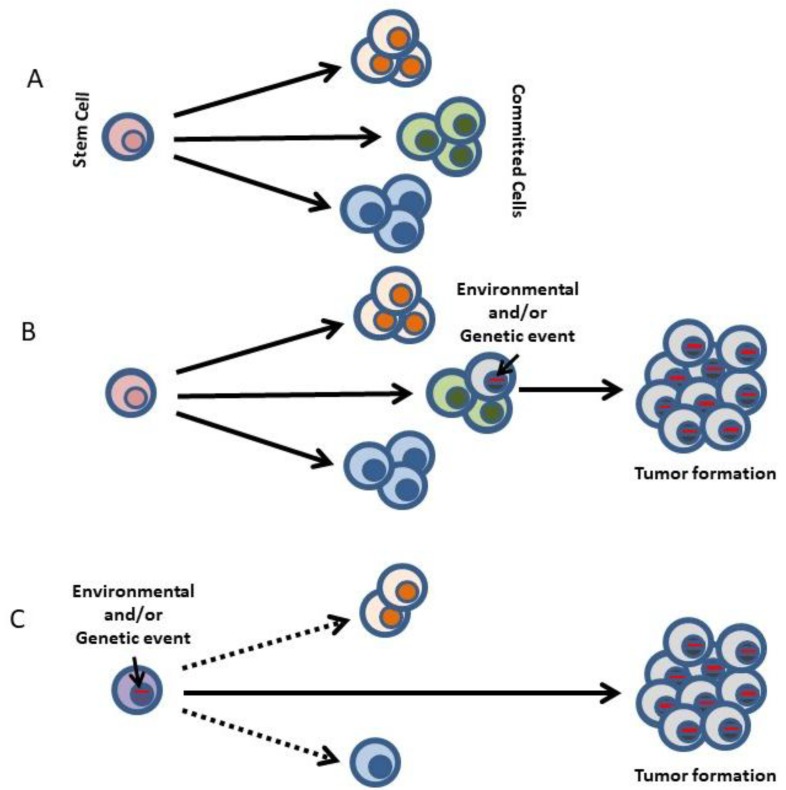
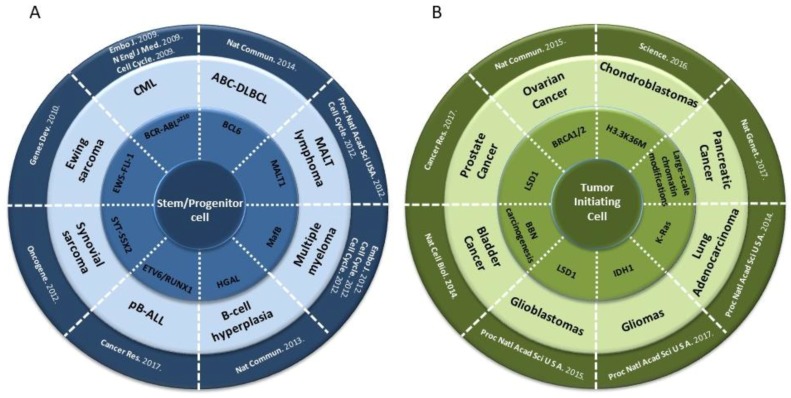
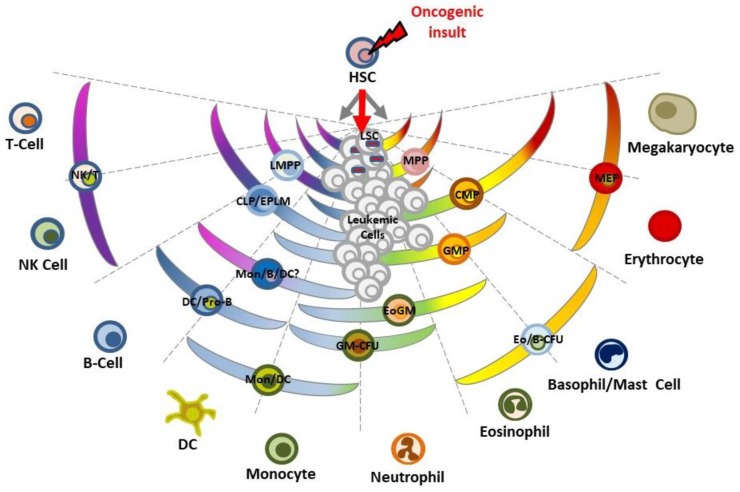
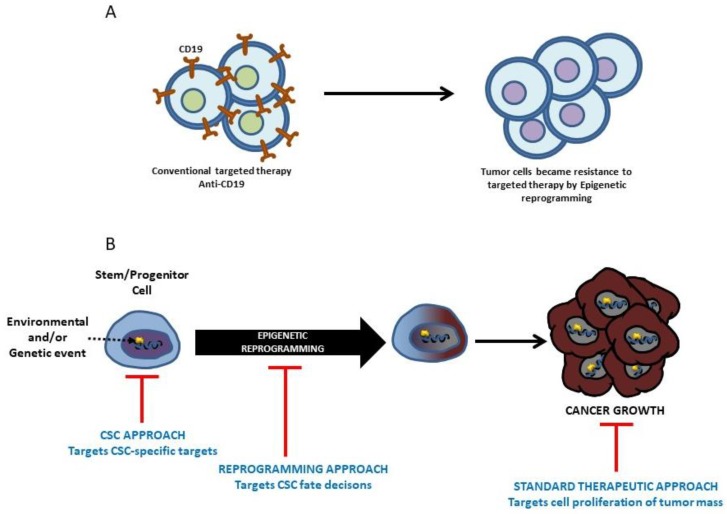
Due to the clonal nature of human leukemia evolution, all leukemic cells carry the same leukemia-initiating genetic lesions, independently of the intrinsic tumoral cellular heterogeneity. However, the latest findings have shown that the mode of action of oncogenes is not homogeneous throughout the developmental history of leukemia. Studies on different types of hematopoietic tumors have shown that the contribution of oncogenes to leukemia is mainly mediated through the epigenetic reprogramming of the leukemia-initiating target cell. This driving of cancer by a malignant epigenetic stem cell rewiring is, however, not exclusive of the hematopoietic system, but rather represents a common tumoral mechanism that is also at work in epithelial tumors. Tumoral epigenetic reprogramming is therefore a new type of interaction between genes and their target cells, in which the action of the oncogene modifies the epigenome to prime leukemia development by establishing a new pathological tumoral cellular identity. This reprogramming may remain latent until it is triggered by either endogenous or environmental stimuli. This new view on the making of leukemia not only reveals a novel function for oncogenes, but also provides evidence for a previously unconsidered model of leukemogenesis, in which the programming of the leukemia cellular identity has already occurred at the level of stem cells, therefore showing a role for oncogenes in the timing of leukemia initiation.
Keywords: cancer therapy; leukemia; leukemia stem cell; mouse model; oncogenes; reprogramming; stem cells.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Rebbeck T.R., Friebel T., Lynch H.T., Neuhausen S.L., van’t Veer L., Garber J.E., Evans G.R., Narod S.A., Isaacs C., Matloff E., et al. Bilateral prophylactic mastectomy reduces breast cancer risk in brca1 and brca2 mutation carriers: The prose study group. J. Clin. Oncol. 2004;22:1055–1062. doi: 10.1200/JCO.2004.04.188. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
