

A thumbwheel mechanism for APOA1 activation of LCAT activity in HDL
- PMID: 29773713
- PMCID: PMC6027914
- DOI: 10.1194/jlr.M085332
A thumbwheel mechanism for APOA1 activation of LCAT activity in HDL
Abstract
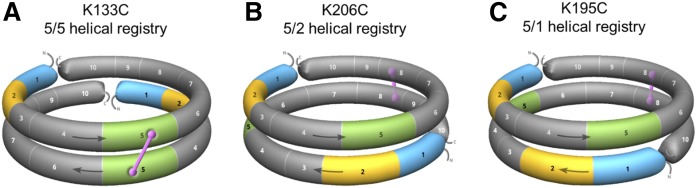
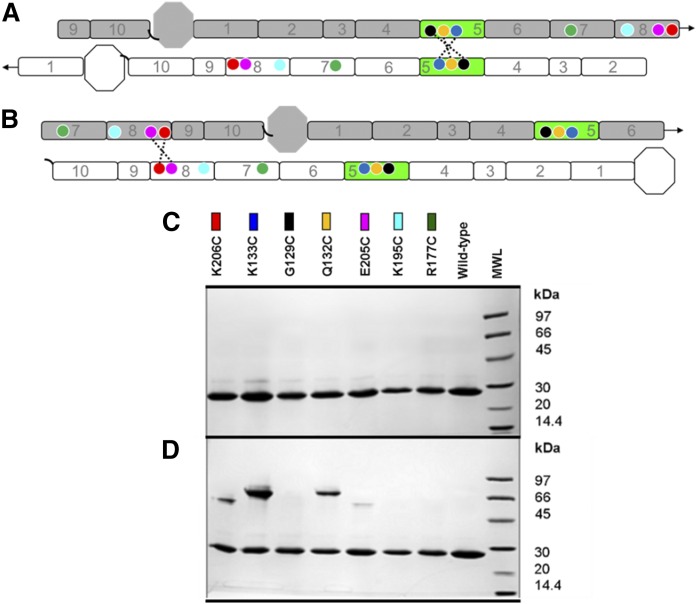
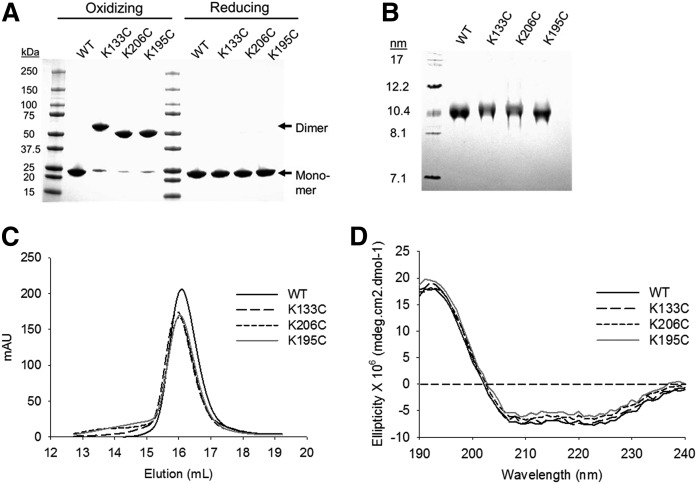
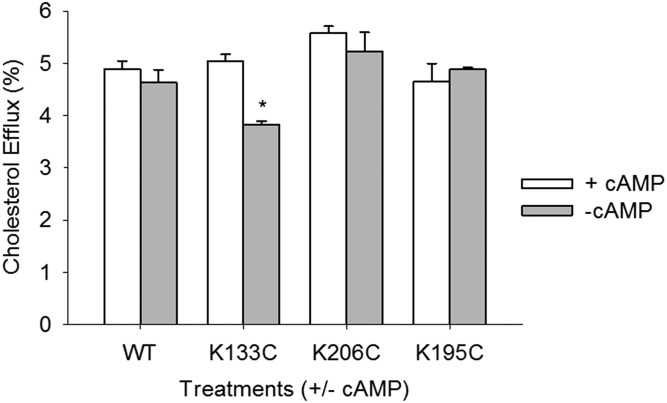
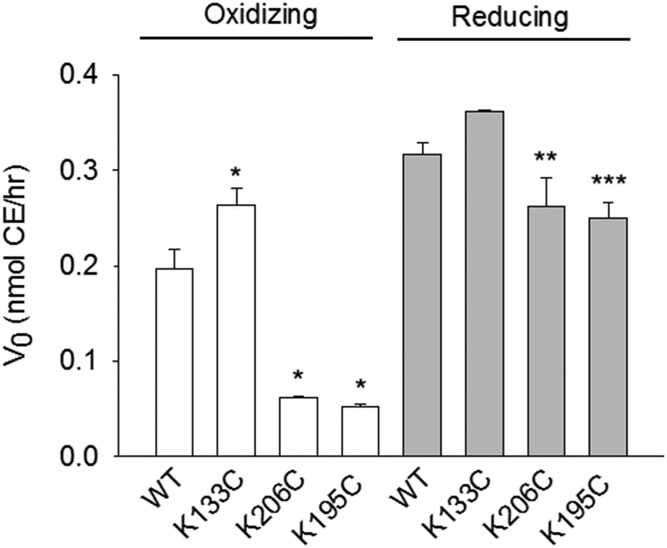
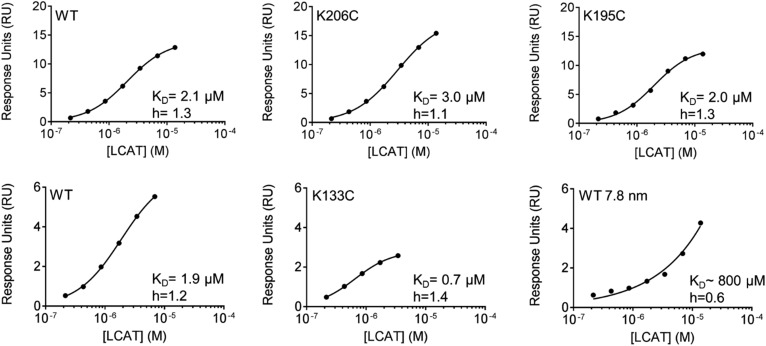
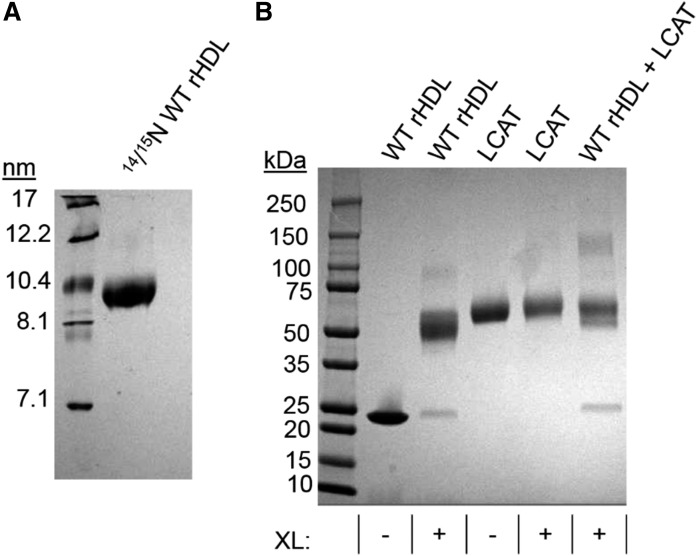
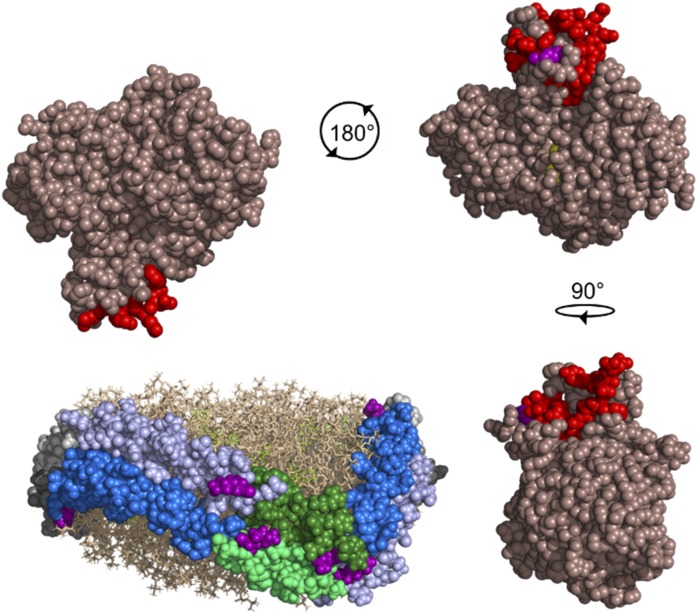
APOA1 is the most abundant protein in HDL. It modulates interactions that affect HDL's cardioprotective functions, in part via its activation of the enzyme, LCAT. On nascent discoidal HDL, APOA1 comprises 10 α-helical repeats arranged in an anti-parallel stacked-ring structure that encapsulates a lipid bilayer. Previous chemical cross-linking studies suggested that these APOA1 rings can adopt at least two different orientations, or registries, with respect to each other; however, the functional impact of these structural changes is unknown. Here, we placed cysteine residues at locations predicted to form disulfide bonds in each orientation and then measured APOA1's ability to adopt the two registries during HDL particle formation. We found that most APOA1 oriented with the fifth helix of one molecule across from fifth helix of the other (5/5 helical registry), but a fraction adopted a 5/2 registry. Engineered HDLs that were locked in 5/5 or 5/2 registries by disulfide bonds equally promoted cholesterol efflux from macrophages, indicating functional particles. However, unlike the 5/5 registry or the WT, the 5/2 registry impaired LCAT cholesteryl esterification activity (P < 0.001), despite LCAT binding equally to all particles. Chemical cross-linking studies suggest that full LCAT activity requires a hybrid epitope composed of helices 5-7 on one APOA1 molecule and helices 3-4 on the other. Thus, APOA1 may use a reciprocating thumbwheel-like mechanism to activate HDL-remodeling proteins.
Keywords: apolipoprotein A1; apolipoproteins; cholesterol metabolism; cholesterol/efflux; electron microscopy; high density lipoprotein; high density lipoprotein metabolism; lecithin:cholesterol acyltransferase; proteomics; surface plasmon resonance.
Conflict of interest statement
The authors declare no financial conflicts of interest.
Figures
References
-
- Gordon T., Castelli W. P., Hjortland M. C., Kannel W. B., and Dawber T. R.. 1977. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med. 62: 707–714. - PubMed
-
- Gu X., Trigatti B., Xu S., Acton S., Babitt J., and Krieger M.. 1998. The efficient cellular uptake of high density lipoprotein lipids via scavenger receptor class B type I requires not only receptor-mediated surface binding but also receptor-specific lipid transfer mediated by its extracellular domain. J. Biol. Chem. 273: 26338–26348. - PubMed
-
- Puranik R., Bao S., Nobecourt E., Nicholls S. J., Dusting G. J., Barter P. J., Celermajer D. S., and Rye K. A.. 2008. Low dose apolipoprotein A-I rescues carotid arteries from inflammation in vivo. Atherosclerosis. 196: 240–247. - PubMed
-
- Navab M., Imes S. S., Hama S. Y., Hough G. P., Ross L. A., Bork R. W., Valente A. J., Berliner J. A., Drinkwater D. C., and Laks H.. 1991. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J. Clin. Invest. 88: 2039–2046. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
