

Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes
- PMID: 29773832
- PMCID: PMC5958058
- DOI: 10.1038/s41467-018-04383-6
Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes
Abstract
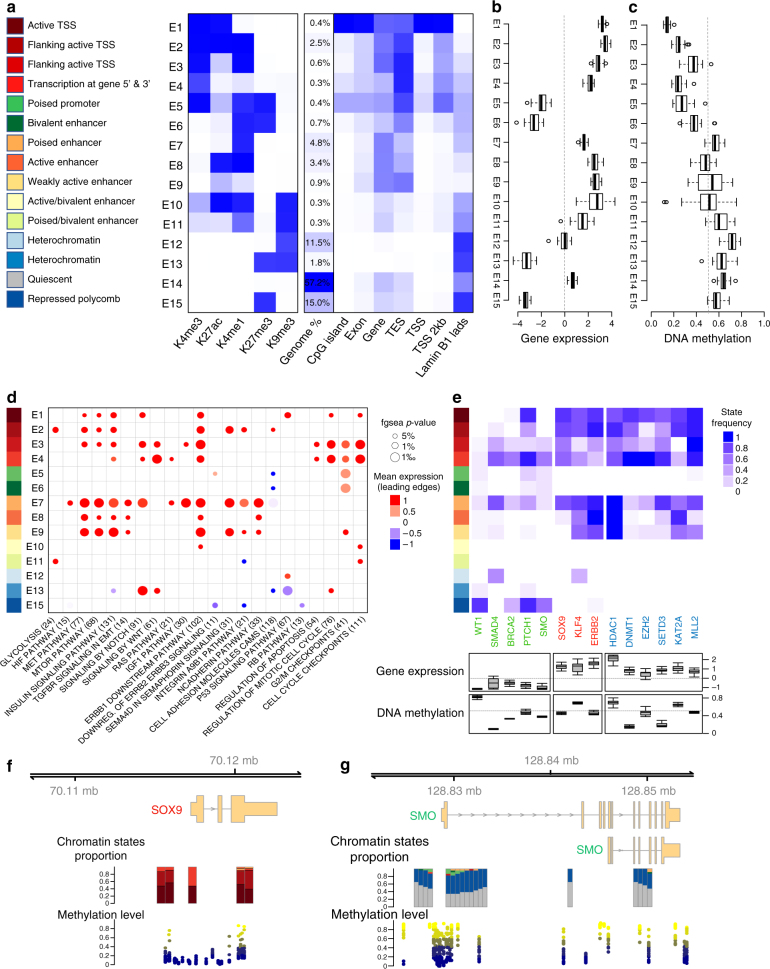
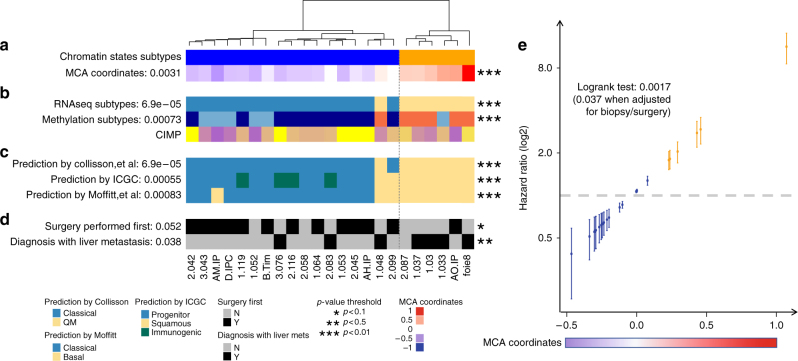
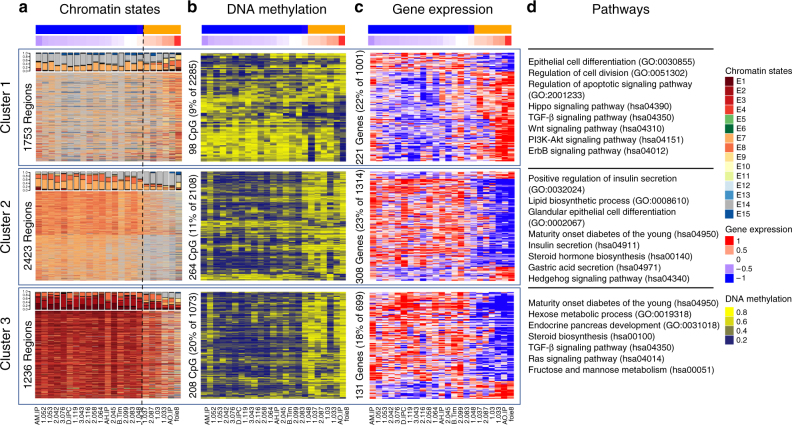
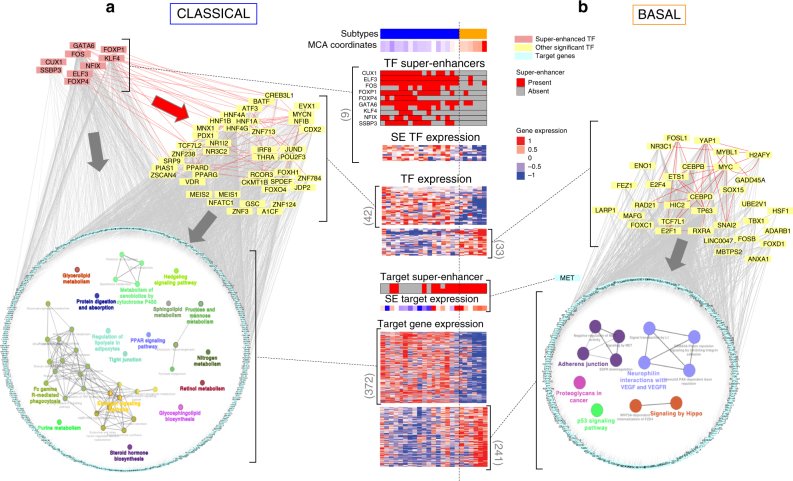
Recent studies have offered ample insight into genome-wide expression patterns to define pancreatic ductal adenocarcinoma (PDAC) subtypes, although there remains a lack of knowledge regarding the underlying epigenomics of PDAC. Here we perform multi-parametric integrative analyses of chromatin immunoprecipitation-sequencing (ChIP-seq) on multiple histone modifications, RNA-sequencing (RNA-seq), and DNA methylation to define epigenomic landscapes for PDAC subtypes, which can predict their relative aggressiveness and survival. Moreover, we describe the state of promoters, enhancers, super-enhancers, euchromatic, and heterochromatic regions for each subtype. Further analyses indicate that the distinct epigenomic landscapes are regulated by different membrane-to-nucleus pathways. Inactivation of a basal-specific super-enhancer associated pathway reveals the existence of plasticity between subtypes. Thus, our study provides new insight into the epigenetic landscapes associated with the heterogeneity of PDAC, thereby increasing our mechanistic understanding of this disease, as well as offering potential new markers and therapeutic targets.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin. Cancer Res. 2000;6:2969. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
