

Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment
- PMID: 29773874
- PMCID: PMC5958119
- DOI: 10.1038/s41467-018-04193-w
Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment
Abstract
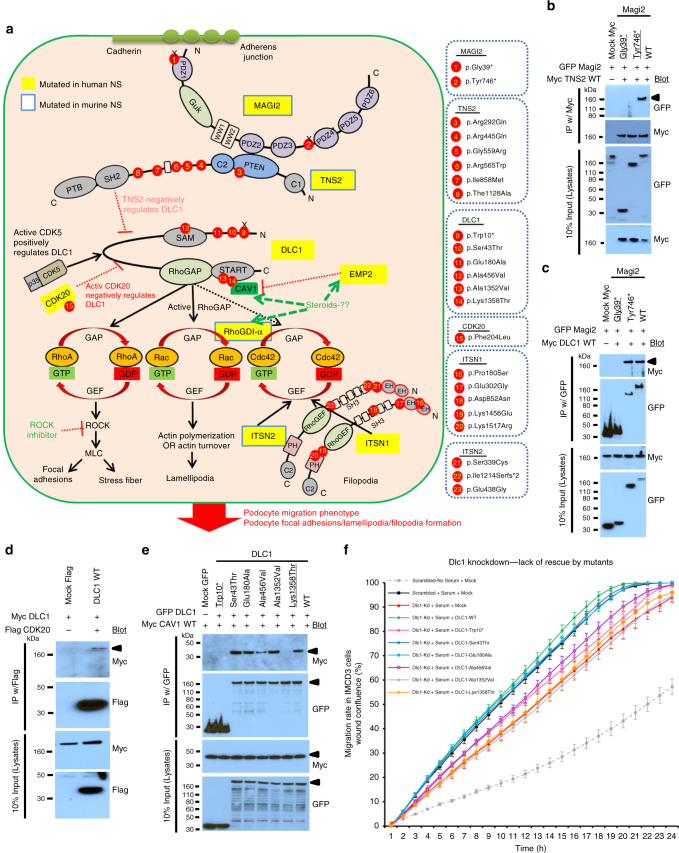
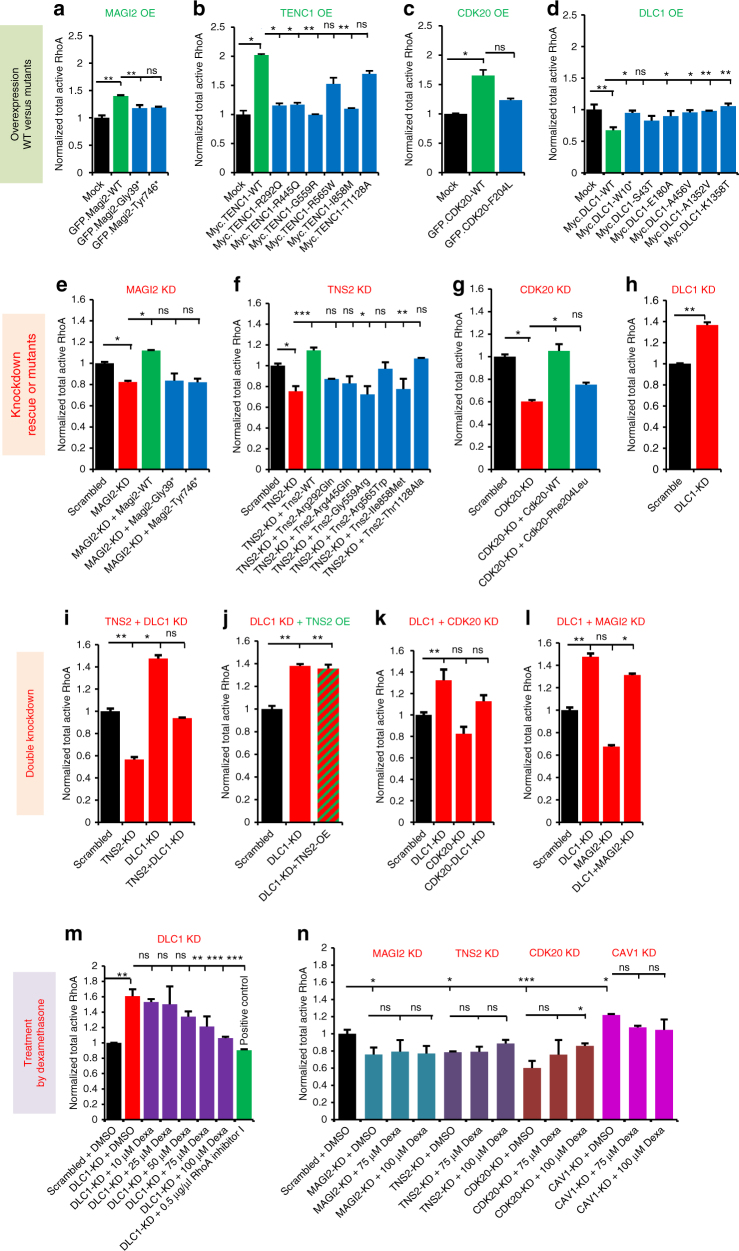
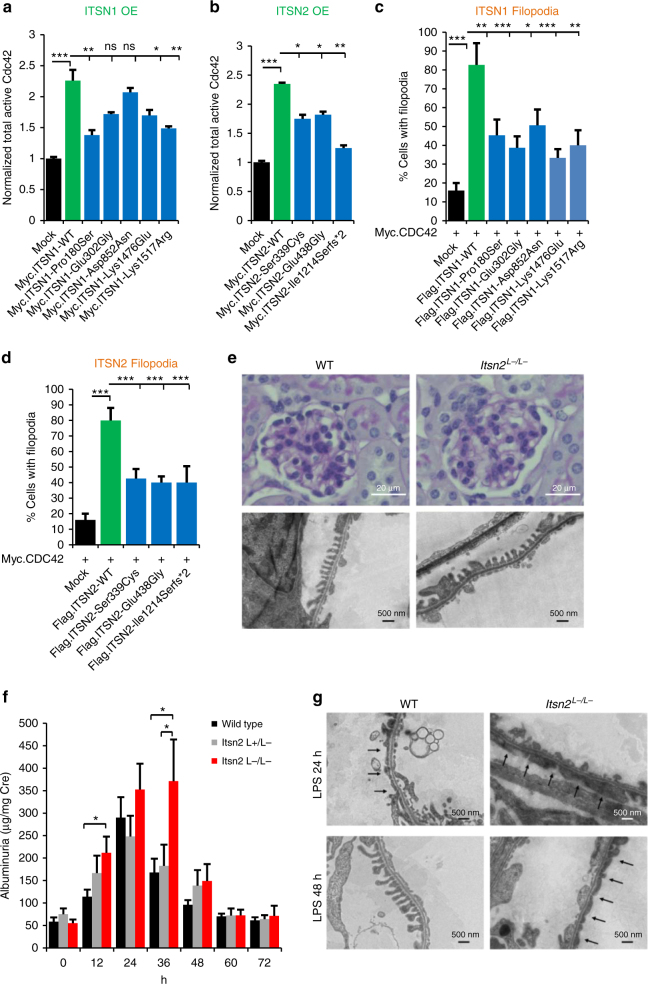
No efficient treatment exists for nephrotic syndrome (NS), a frequent cause of chronic kidney disease. Here we show mutations in six different genes (MAGI2, TNS2, DLC1, CDK20, ITSN1, ITSN2) as causing NS in 17 families with partially treatment-sensitive NS (pTSNS). These proteins interact and we delineate their roles in Rho-like small GTPase (RLSG) activity, and demonstrate deficiency for mutants of pTSNS patients. We find that CDK20 regulates DLC1. Knockdown of MAGI2, DLC1, or CDK20 in cultured podocytes reduces migration rate. Treatment with dexamethasone abolishes RhoA activation by knockdown of DLC1 or CDK20 indicating that steroid treatment in patients with pTSNS and mutations in these genes is mediated by this RLSG module. Furthermore, we discover ITSN1 and ITSN2 as podocytic guanine nucleotide exchange factors for Cdc42. We generate Itsn2-L knockout mice that recapitulate the mild NS phenotype. We, thus, define a functional network of RhoA regulation, thereby revealing potential therapeutic targets.
Conflict of interest statement
F.H. is a cofounder of Goldfinch-Bio. All remaining authors declare no competing interests.
Figures

Comment in
-
RHO-like GTPases in nephrotic syndrome.Nat Rev Nephrol. 2018 Sep;14(9):536. doi: 10.1038/s41581-018-0030-6. Nat Rev Nephrol. 2018. PMID: 29855604 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
