

Evaluation of phylogenetic reconstruction methods using bacterial whole genomes: a simulation based study
- PMID: 29774245
- PMCID: PMC5930550
- DOI: 10.12688/wellcomeopenres.14265.2
Evaluation of phylogenetic reconstruction methods using bacterial whole genomes: a simulation based study
Abstract
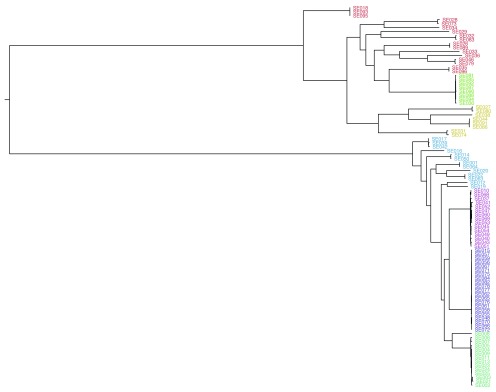
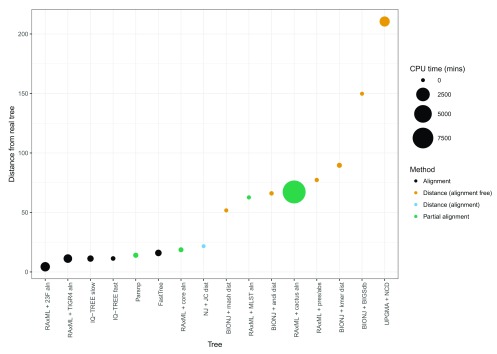
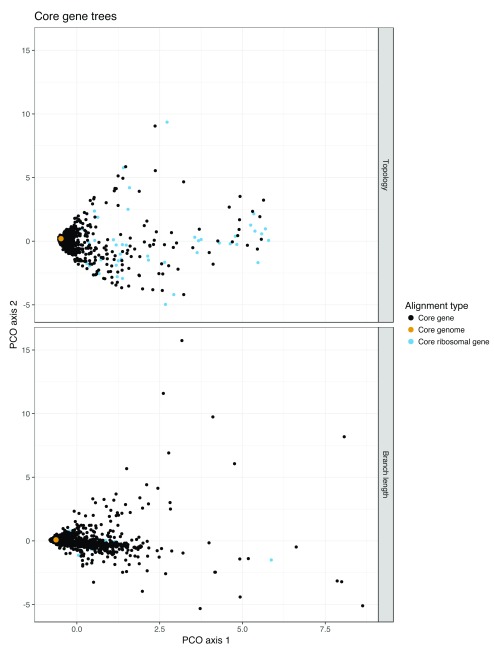
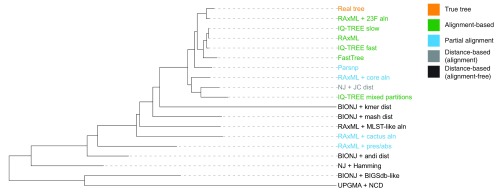
Background: Phylogenetic reconstruction is a necessary first step in many analyses which use whole genome sequence data from bacterial populations. There are many available methods to infer phylogenies, and these have various advantages and disadvantages, but few unbiased comparisons of the range of approaches have been made. Methods: We simulated data from a defined "true tree" using a realistic evolutionary model. We built phylogenies from this data using a range of methods, and compared reconstructed trees to the true tree using two measures, noting the computational time needed for different phylogenetic reconstructions. We also used real data from Streptococcus pneumoniae alignments to compare individual core gene trees to a core genome tree. Results: We found that, as expected, maximum likelihood trees from good quality alignments were the most accurate, but also the most computationally intensive. Using less accurate phylogenetic reconstruction methods, we were able to obtain results of comparable accuracy; we found that approximate results can rapidly be obtained using genetic distance based methods. In real data we found that highly conserved core genes, such as those involved in translation, gave an inaccurate tree topology, whereas genes involved in recombination events gave inaccurate branch lengths. We also show a tree-of-trees, relating the results of different phylogenetic reconstructions to each other. Conclusions: We recommend three approaches, depending on requirements for accuracy and computational time. Quicker approaches that do not perform full maximum likelihood optimisation may be useful for many analyses requiring a phylogeny, as generating a high quality input alignment is likely to be the major limiting factor of accurate tree topology. We have publicly released our simulated data and code to enable further comparisons.
Keywords: bacteria; phylogenetic methods; phylogeny; simulation; tree distance.
Conflict of interest statement
No competing interests were disclosed.
Figures
References
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
