

QM/MM Simulations with the Gaussian Electrostatic Model: A Density-based Polarizable Potential
- PMID: 29775314
- PMCID: PMC6069983
- DOI: 10.1021/acs.jpclett.8b01412
QM/MM Simulations with the Gaussian Electrostatic Model: A Density-based Polarizable Potential
Abstract
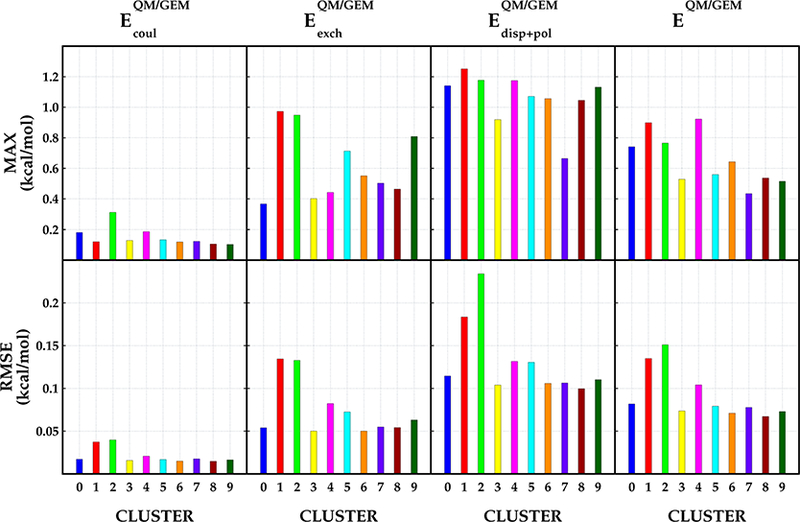
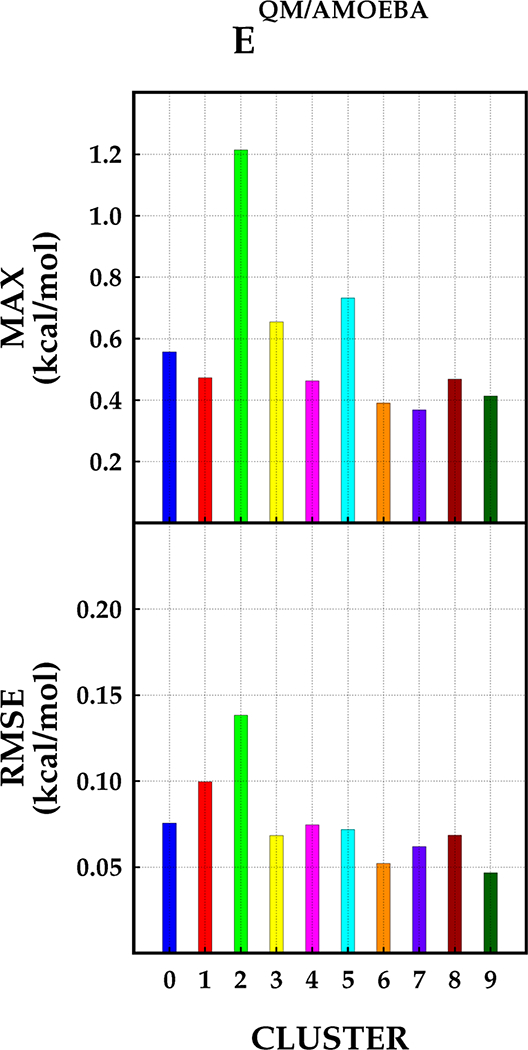
The use of advanced polarizable potentials in quantum mechanical/molecular mechanical (QM/MM) simulations has been shown to improve the overall accuracy of the calculation. We have developed a density-based potential called the Gaussian electrostatic model (GEM), which has been shown to provide very accurate environments for QM wave functions in QM/MM. In this contribution we present a new implementation of QM/GEM that extends our implementation to include all components (Coulomb, exchange-repulsion, polarization, and dispersion) for the total intermolecular interaction energy in QM/MM calculations, except for the charge-transfer term. The accuracy of the method is tested using a subset of water dimers from the water dimer potential energy surface reported by Babin et al. ( J. Chem. Theory Comput. 2013 9, 5395-5403). Additionally, results of the new implementation are contrasted with results obtained with the classical AMOEBA potential. Our results indicate that GEM provides an accurate MM environment with average root-mean-square error <0.15 kcal/mol for every intermolecular interaction energy component compared with SAPT2+3/aug-cc-pVTZ reference calculations.
Conflict of interest statement
Notes
The authors declare no competing financial interest.
Figures
Similar articles
-
Accurate description of intermolecular interactions involving ions using symmetry-adapted perturbation theory.J Chem Theory Comput. 2015 Jun 9;11(6):2473-86. doi: 10.1021/ct5010593. Epub 2015 May 4. J Chem Theory Comput. 2015. PMID: 26575547
-
A fixed multi-site interaction charge model for an accurate prediction of the QM/MM interactions.Phys Chem Chem Phys. 2021 Sep 29;23(37):21001-21012. doi: 10.1039/d1cp02776j. Phys Chem Chem Phys. 2021. PMID: 34522933
-
Generalization of the Gaussian electrostatic model: extension to arbitrary angular momentum, distributed multipoles, and speedup with reciprocal space methods.J Chem Phys. 2006 Nov 14;125(18):184101. doi: 10.1063/1.2363374. J Chem Phys. 2006. PMID: 17115732 Free PMC article.
-
Seamless integration of GEM, a density based-force field, for QM/MM simulations via LICHEM, Psi4, and Tinker-HP.J Chem Phys. 2024 May 7;160(17):174103. doi: 10.1063/5.0200722. J Chem Phys. 2024. PMID: 38747990 Free PMC article.
-
Current Status of AMOEBA-IL: A Multipolar/Polarizable Force Field for Ionic Liquids.Int J Mol Sci. 2020 Jan 21;21(3):697. doi: 10.3390/ijms21030697. Int J Mol Sci. 2020. PMID: 31973103 Free PMC article. Review.
Cited by
-
Development and application of quantum mechanics/molecular mechanics methods with advanced polarizable potentials.Wiley Interdiscip Rev Comput Mol Sci. 2021 Jul-Aug;11(4):e1515. doi: 10.1002/wcms.1515. Epub 2021 Jan 12. Wiley Interdiscip Rev Comput Mol Sci. 2021. PMID: 34367343 Free PMC article.
-
A simplified charge projection scheme for long-range electrostatics in ab initio QM/MM calculations.J Chem Phys. 2021 Jan 14;154(2):024115. doi: 10.1063/5.0038120. J Chem Phys. 2021. PMID: 33445891 Free PMC article.
-
A Simple Electron-Density Based Force Field Model for High-Energy Interactions between Atoms and Molecules.J Phys Chem A. 2024 Feb 15;128(6):1163-1172. doi: 10.1021/acs.jpca.3c06724. Epub 2024 Feb 6. J Phys Chem A. 2024. PMID: 38320398 Free PMC article.
-
Intermolecular Non-Bonded Interactions from Machine Learning Datasets.Molecules. 2023 Dec 1;28(23):7900. doi: 10.3390/molecules28237900. Molecules. 2023. PMID: 38067629 Free PMC article.
-
Modeling UV/Vis Absorption Spectra of Food Colorants in Solution: Anthocyanins and Curcumin as Case Studies.Molecules. 2024 Sep 14;29(18):4378. doi: 10.3390/molecules29184378. Molecules. 2024. PMID: 39339373 Free PMC article.
References
-
- Senn H; Thiel W QM/MM Methods for Biological Systems In Atomistic Approaches in Modern Biology; Topics in Current Chemistry 268; Springer: Berlin, 2007; pp 173–290.
-
- Senn HM; Thiel W QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed 2009, 48, 1198–1229. - PubMed
-
- van der Kamp MW; Mulholland AJ Combined Quantum Mechanics/Molecular Mechanics (QM/MM) Methods in Computational Enzymology. Biochemistry 2013, 52, 2708–2728. - PubMed
-
- Claeyssens F; Ranaghan KE; Manby FR; Harvey JN; Mulholland AJ Multiple high-level QM/MM reaction paths demonstrate transition-state stabilization in chorismate mutase: correlation of barrier height with transition-state stabilization. Chem. Commun 2005, 5068–5070. - PubMed
-
- Woods CJ; Manby FR; Mulholland AJ An efficient method for the calculation of quantum mechanics/molecular mechanics free energies. J. Chem. Phys 2008, 128, 014109. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
