

Novel peptide nanoparticle-biased antagonist of CCR3 blocks eosinophil recruitment and airway hyperresponsiveness
- PMID: 29778505
- PMCID: PMC6240402
- DOI: 10.1016/j.jaci.2018.05.003
Novel peptide nanoparticle-biased antagonist of CCR3 blocks eosinophil recruitment and airway hyperresponsiveness
Abstract
Background: Chemokine signaling through CCR3 is a key regulatory pathway for eosinophil recruitment into tissues associated with allergic inflammation and asthma. To date, none of the CCR3 antagonists have shown efficacy in clinical trials. One reason might be their unbiased mode of inhibition that prevents receptor internalization, leading to drug tolerance.
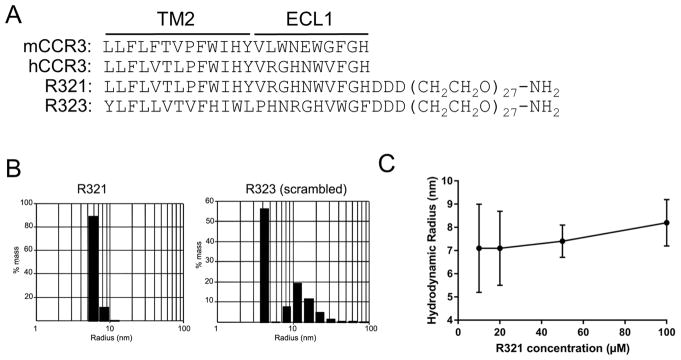
Objective: We sought to develop a novel peptide nanoparticle CCR3 inhibitor (R321) with a biased mode of inhibition that would block G protein signaling but enable or promote receptor internalization.
Methods: Self-assembly of R321 peptide into nanoparticles and peptide binding to CCR3 were analyzed by means of dynamic light scattering and nuclear magnetic resonance. Inhibitory activity on CCR3 signaling was assessed in vitro by using flow cytometry, confocal microscopy, and Western blot analysis in a CCR3+ eosinophil cell line and blood eosinophils. In vivo effects of R321 were assessed by using a triple-allergen mouse asthma model.
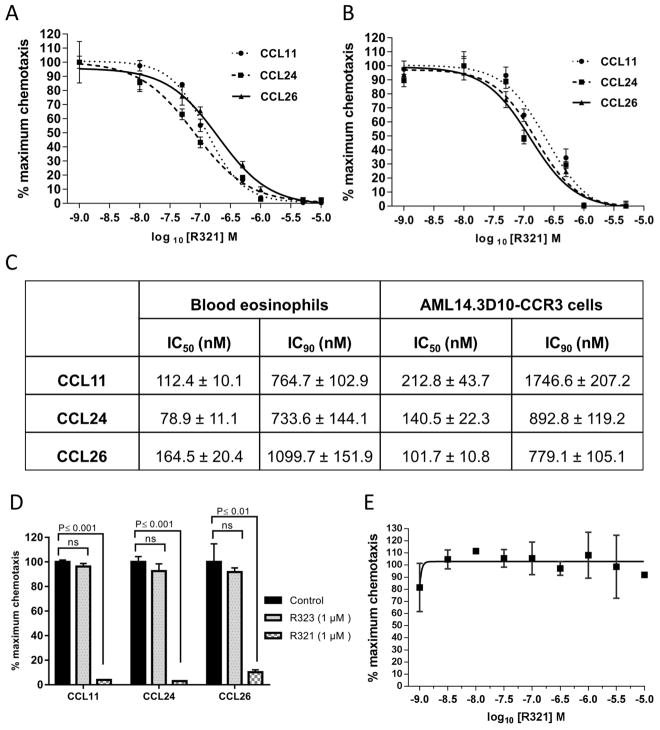
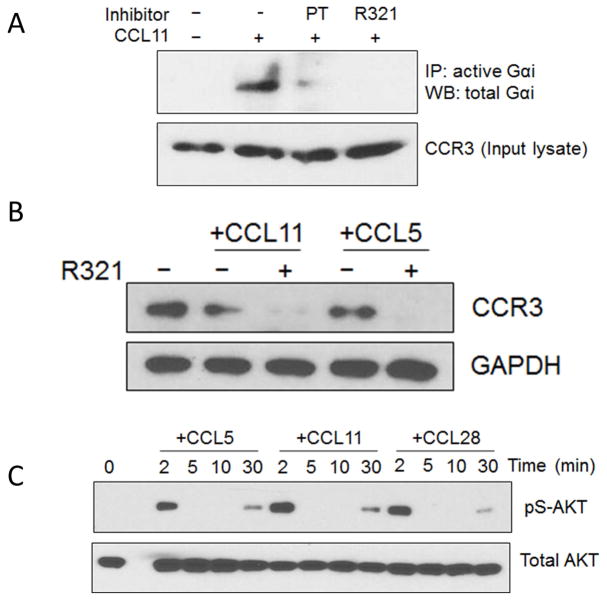
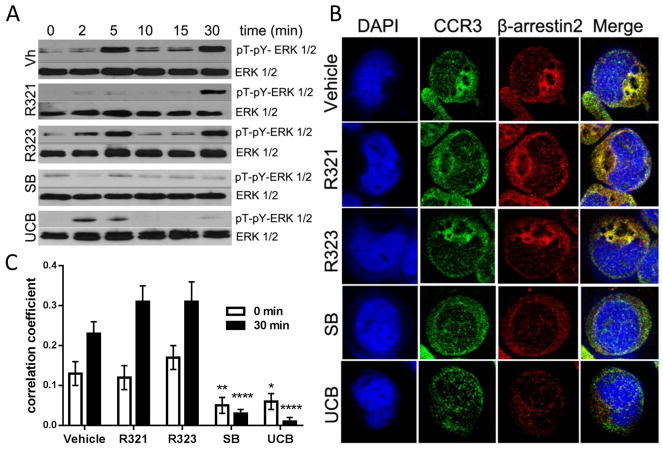
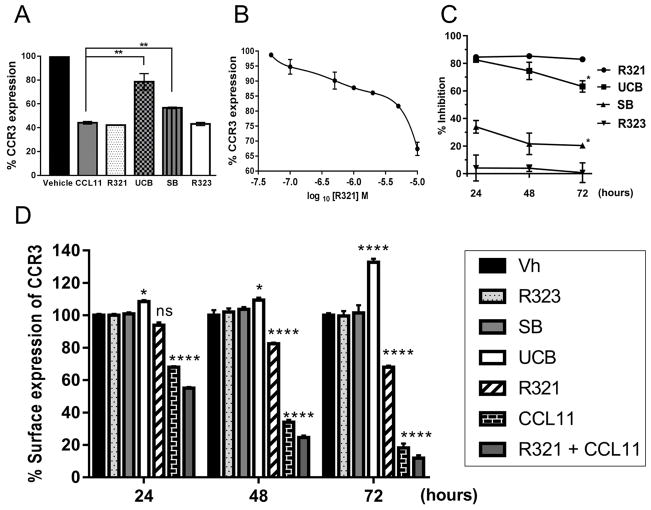
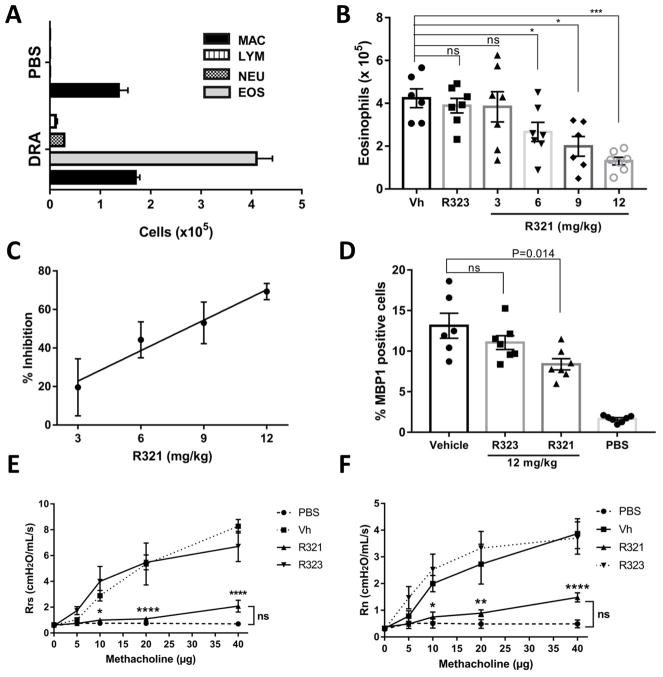
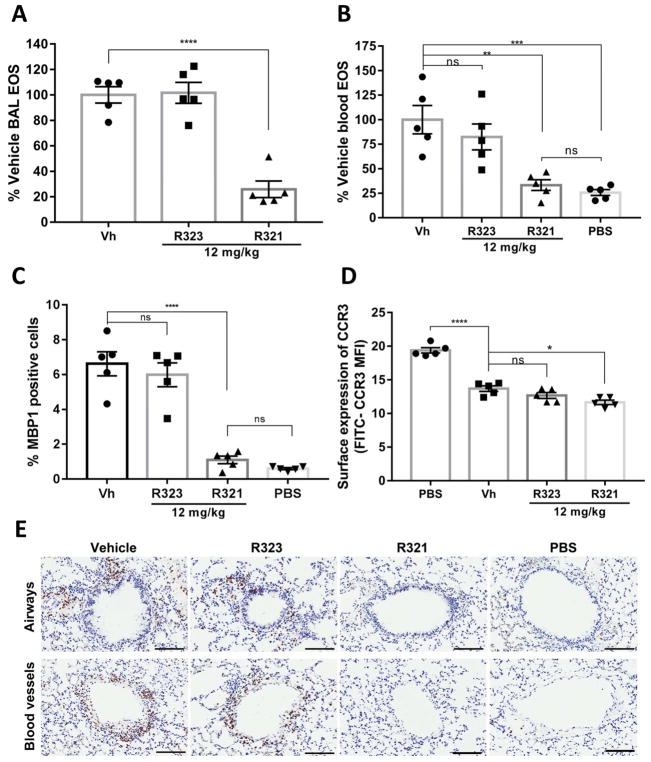
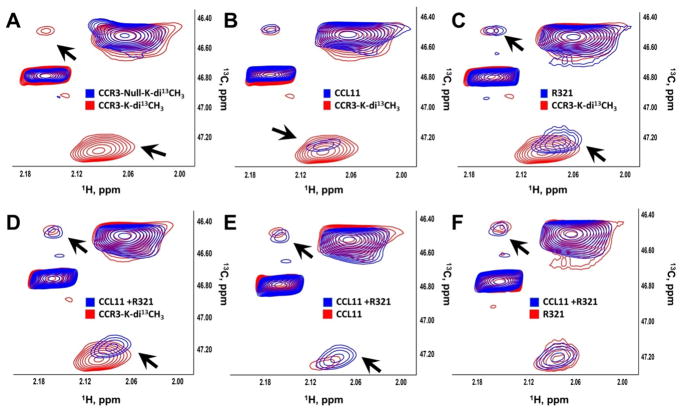
Results: R321 self-assembles into nanoparticles and binds directly to CCR3, altering receptor function. Half-maximal inhibitory concentration values for eotaxin-induced chemotaxis of blood eosinophils are in the low nanomolar range. R321 inhibits only the early phase of extracellular signal-regulated kinase 1/2 activation and not the late phase generally associated with β-arrestin recruitment and receptor endocytosis, promoting CCR3 internalization and degradation. In vivo R321 effectively blocks eosinophil recruitment into the blood, lungs, and airways and prevents airway hyperresponsiveness in a mouse eosinophilic asthma model.
Conclusions: R321 is a potent and selective antagonist of the CCR3 signaling cascade. Inhibition through a biased mode of antagonism might hold significant therapeutic promise by eluding the formation of drug tolerance.
Keywords: CCR3; airway hyperresponsiveness; allergic inflammation; asthma; biased antagonist; eosinophil; peptide nanoparticles.
Copyright © 2018 American Academy of Allergy, Asthma & Immunology. All rights reserved.
Conflict of interest statement
Figures
Comment in
-
Tipping the balance: A biased nanobody antagonist of CCR3 with potential for the treatment of eosinophilic inflammation.J Allergy Clin Immunol. 2019 Feb;143(2):552-553. doi: 10.1016/j.jaci.2018.10.052. Epub 2018 Nov 17. J Allergy Clin Immunol. 2019. PMID: 30452926 No abstract available.
References
-
- Busse W, Sedgwick J. Eosinophils in asthma. Annals of Allergy. 1992;68:286–90. - PubMed
-
- Markowitz JE, Liacouras CA. Eosinophilic esophagitis. Gastroenterology Clinics of North America. 2003;32:949–66. - PubMed
-
- Willems LI, Ijzerman AP. Small molecule antagonists for chemokine CCR3 receptors. Medicinal research reviews. 2010;30:778–817. - PubMed
-
- Pease JE, Horuk R. Recent progress in the development of antagonists to the chemokine receptors CCR3 and CCR4. Expert opinion on drug discovery. 2014;9:467–83. - PubMed
-
- Stellato C, Brummet M, Plitt J, Shahabuddin S, Baroody F, Liu M, et al. Expression of the C-C chemokine receptor CCR3 in human airway epithelial cells. Journal of Immunology. 2001;166:1457–61. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
