

Hereditary sensory neuropathy type 1-associated deoxysphingolipids cause neurotoxicity, acute calcium handling abnormalities and mitochondrial dysfunction in vitro
- PMID: 29778900
- PMCID: PMC6060082
- DOI: 10.1016/j.nbd.2018.05.008
Hereditary sensory neuropathy type 1-associated deoxysphingolipids cause neurotoxicity, acute calcium handling abnormalities and mitochondrial dysfunction in vitro
Abstract
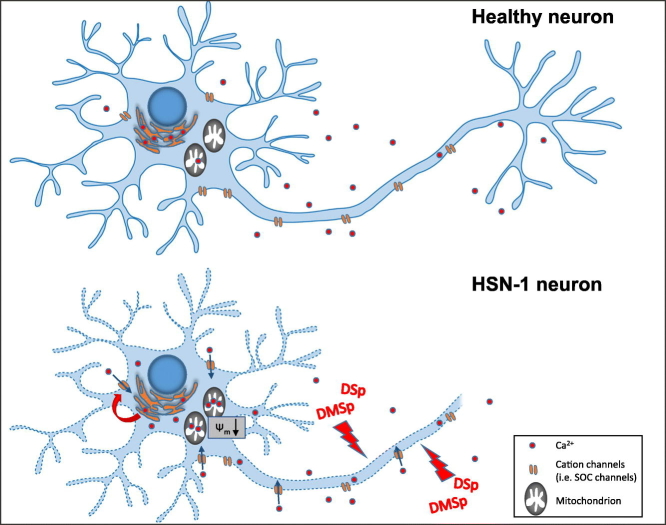
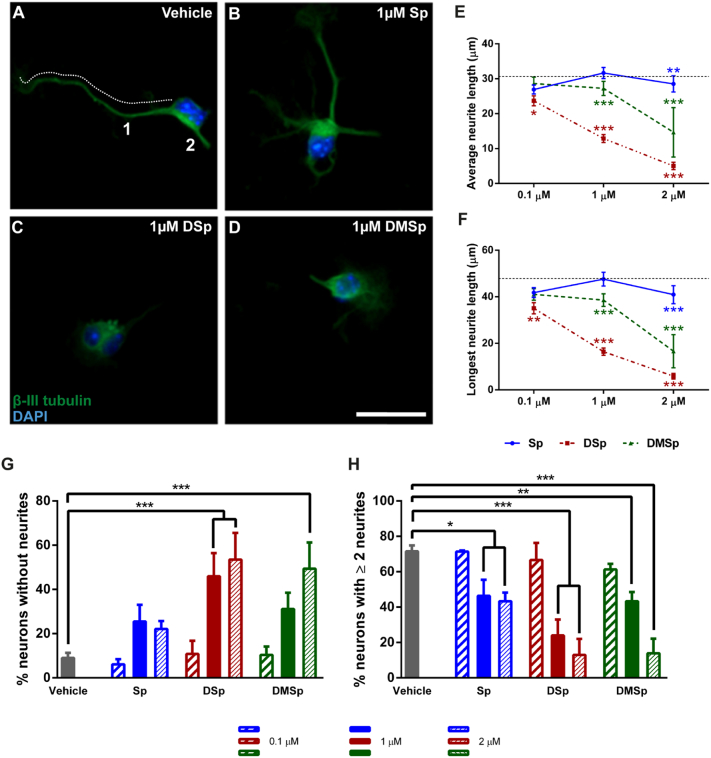
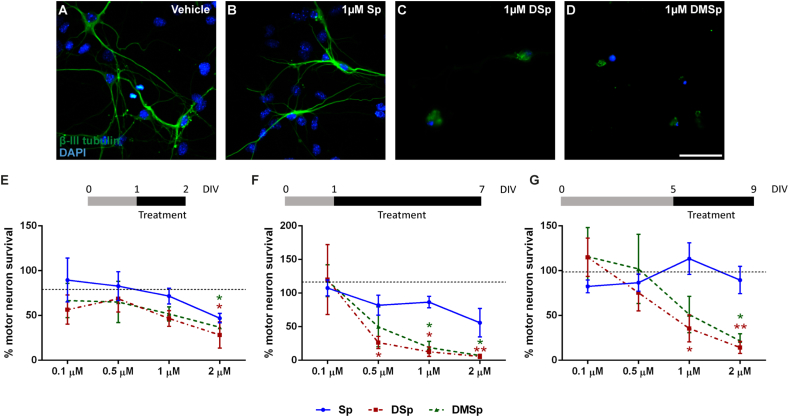
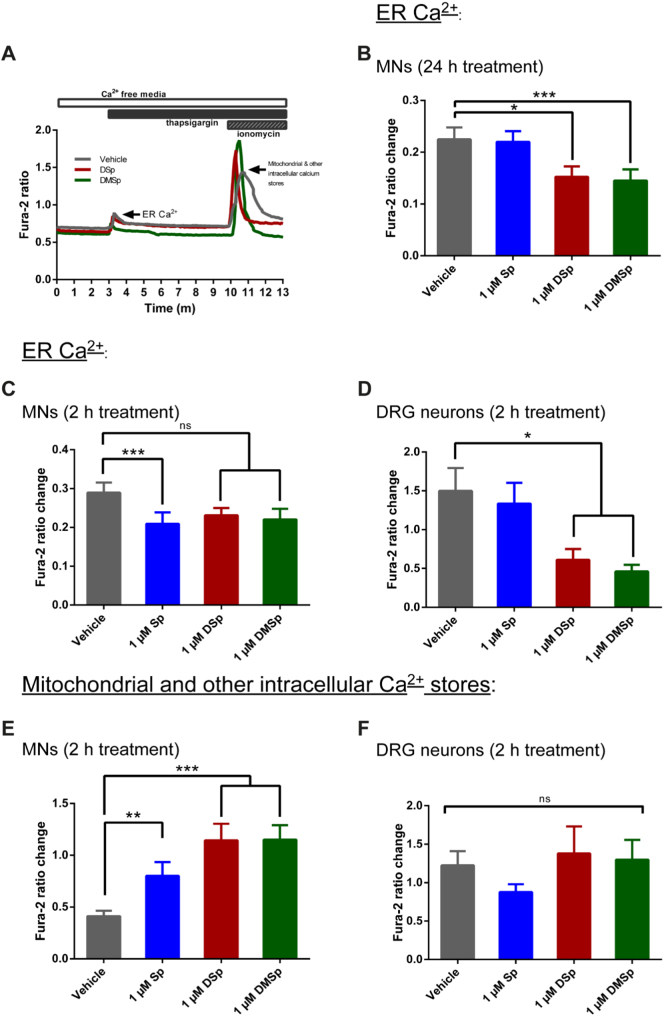
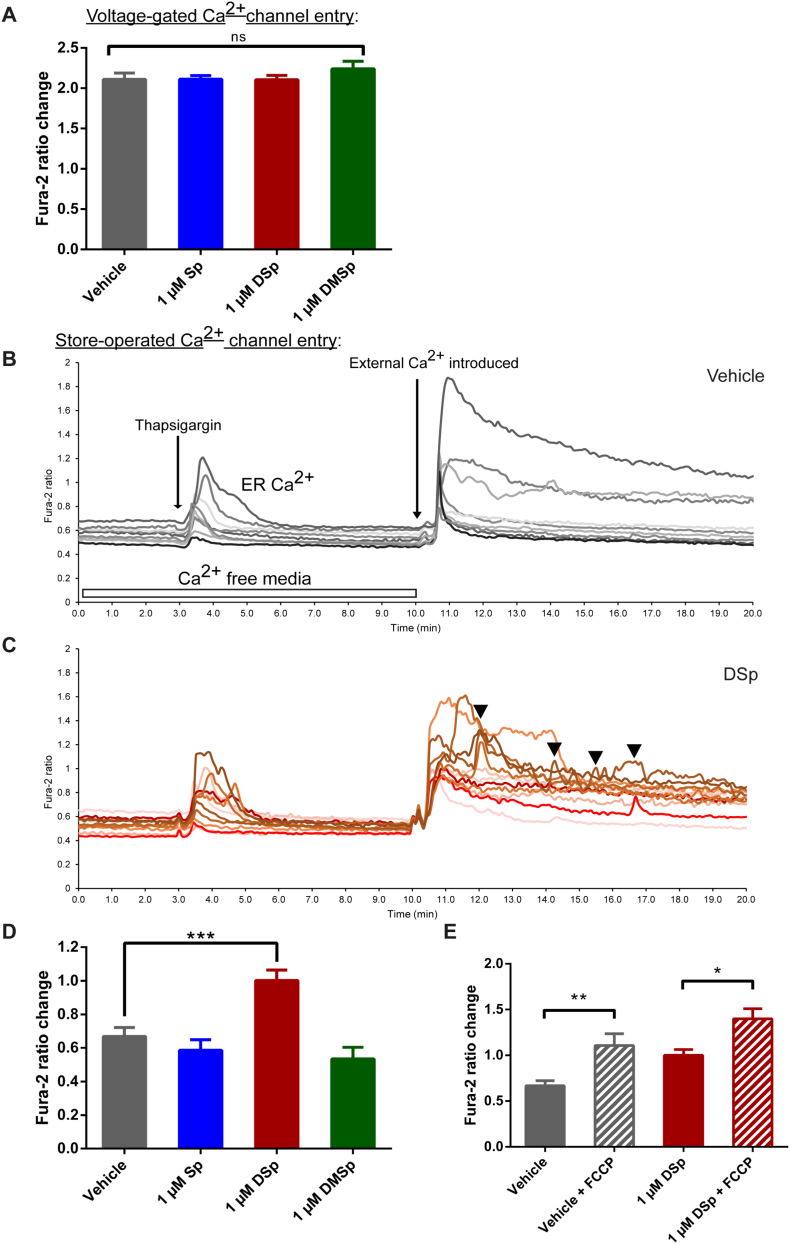
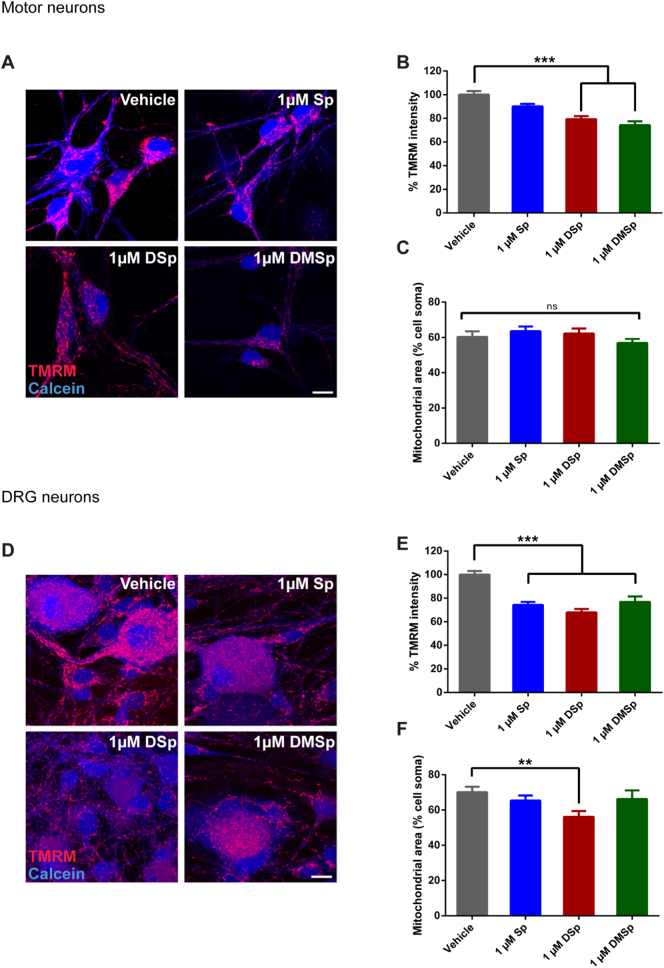
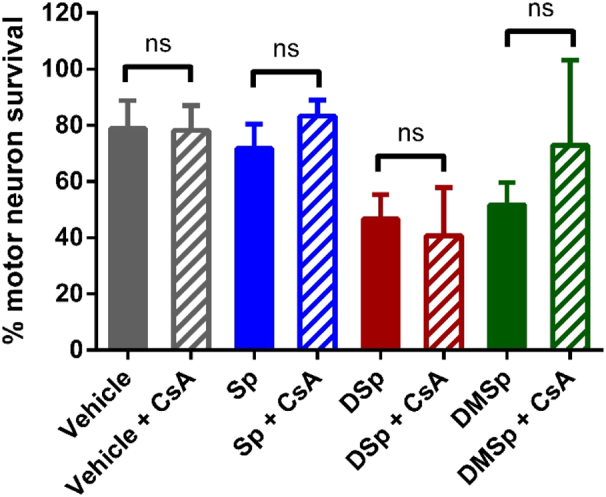
Hereditary sensory neuropathy type 1 (HSN-1) is a peripheral neuropathy most frequently caused by mutations in the SPTLC1 or SPTLC2 genes, which code for two subunits of the enzyme serine palmitoyltransferase (SPT). SPT catalyzes the first step of de novo sphingolipid synthesis. Mutations in SPT result in a change in enzyme substrate specificity, which causes the production of atypical deoxysphinganine and deoxymethylsphinganine, rather than the normal enzyme product, sphinganine. Levels of these abnormal compounds are elevated in blood of HSN-1 patients and this is thought to cause the peripheral motor and sensory nerve damage that is characteristic of the disease, by a largely unresolved mechanism. In this study, we show that exogenous application of these deoxysphingoid bases causes dose- and time-dependent neurotoxicity in primary mammalian neurons, as determined by analysis of cell survival and neurite length. Acutely, deoxysphingoid base neurotoxicity manifests in abnormal Ca2+ handling by the endoplasmic reticulum (ER) and mitochondria as well as dysregulation of cell membrane store-operated Ca2+ channels. The changes in intracellular Ca2+ handling are accompanied by an early loss of mitochondrial membrane potential in deoxysphingoid base-treated motor and sensory neurons. Thus, these results suggest that exogenous deoxysphingoid base application causes neuronal mitochondrial dysfunction and Ca2+ handling deficits, which may play a critical role in the pathogenesis of HSN-1.
Keywords: Deoxysphingolipid; ES-285; Endoplasmic reticulum; Mitochondria; Neuron; Peripheral neuropathy; Sphingolipid.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Characterization of two mutations in the SPTLC1 subunit of serine palmitoyltransferase associated with hereditary sensory and autonomic neuropathy type I.Hum Mutat. 2011 Jun;32(6):E2211-25. doi: 10.1002/humu.21481. Epub 2011 Feb 24. Hum Mutat. 2011. PMID: 21618344
-
Serine and Lipid Metabolism in Macular Disease and Peripheral Neuropathy.N Engl J Med. 2019 Oct 10;381(15):1422-1433. doi: 10.1056/NEJMoa1815111. Epub 2019 Sep 11. N Engl J Med. 2019. PMID: 31509666 Free PMC article.
-
Demyelination in hereditary sensory neuropathy type-1C.Ann Clin Transl Neurol. 2020 Sep;7(9):1502-1512. doi: 10.1002/acn3.51110. Epub 2020 Jul 30. Ann Clin Transl Neurol. 2020. PMID: 32730653 Free PMC article.
-
1-Deoxysphingolipids Encountered Exogenously and Made de Novo: Dangerous Mysteries inside an Enigma.J Biol Chem. 2015 Jun 19;290(25):15380-15389. doi: 10.1074/jbc.R115.658823. Epub 2015 May 6. J Biol Chem. 2015. PMID: 25947379 Free PMC article. Review.
-
Serine Palmitoyltransferase (SPT)-related Neurodegenerative and Neurodevelopmental Disorders.J Neuromuscul Dis. 2024;11(4):735-747. doi: 10.3233/JND-240014. J Neuromuscul Dis. 2024. PMID: 38788085 Free PMC article. Review.
Cited by
-
Neurological update: hereditary neuropathies.J Neurol. 2022 Sep;269(9):5187-5191. doi: 10.1007/s00415-022-11164-1. Epub 2022 May 21. J Neurol. 2022. PMID: 35596796 Free PMC article. Review.
-
Imbalanced unfolded protein response signaling contributes to 1-deoxysphingolipid retinal toxicity.Nat Commun. 2023 Jul 11;14(1):4119. doi: 10.1038/s41467-023-39775-w. Nat Commun. 2023. PMID: 37433773 Free PMC article.
-
Metabolism of HSAN1- and T2DM-associated 1-deoxy-sphingolipids inhibits the migration of fibroblasts.J Lipid Res. 2021;62:100122. doi: 10.1016/j.jlr.2021.100122. Epub 2021 Sep 24. J Lipid Res. 2021. PMID: 34563520 Free PMC article.
-
Genetic pain loss disorders.Nat Rev Dis Primers. 2022 Jun 16;8(1):41. doi: 10.1038/s41572-022-00365-7. Nat Rev Dis Primers. 2022. PMID: 35710757 Review.
-
Human genetic defects of sphingolipid synthesis.J Inherit Metab Dis. 2025 Jan;48(1):e12745. doi: 10.1002/jimd.12745. Epub 2024 May 5. J Inherit Metab Dis. 2025. PMID: 38706107 Free PMC article. Review.
References
-
- Abramov A.Y., Duchen M.R. Actions of ionomycin, 4-BrA23187 and a novel electrogenic Ca2+ ionophore on mitochondria in intact cells. Cell Calcium. 2003;33:101–112. - PubMed
-
- Alecu I., Tedeschi A., Behler N., Wunderling K., Lamberz C., Lauterbach M.A., Gaebler A., Ernst D., Van Veldhoven P.P., Al-Amoudi A., Latz E., Othman A., Kuerschner L., Hornemann T., Bradke F., Thiele C., Penno A. Localization of 1-deoxysphingolipids to mitochondria induces mitochondrial dysfunction. J. Lipid Res. 2016 - PMC - PubMed
-
- Auer-Grumbach M. Hereditary sensory and autonomic neuropathies. Handb. Clin. Neurol. 2013;115:893–906. - PubMed
-
- Auer-Grumbach M., Bode H., Pieber T.R., Schabhuttl M., Fischer D., Seidl R., Graf E., Wieland T., Schuh R., Vacariu G., Grill F., Timmerman V., Strom T.M., Hornemann T. Mutations at Ser331 in the HSN type I gene SPTLC1 are associated with a distinct syndromic phenotype. Eur. J. Med. Genet. 2013;56:266–269. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
