

Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases
- PMID: 29780380
- PMCID: PMC5946030
- DOI: 10.3389/fimmu.2018.00832
Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases
Abstract
Over the recent years, much has been unraveled about the pro-inflammatory properties of various mitochondrial molecules once they are leaving the mitochondrial compartment. On entering the cytoplasm or the extracellular space, mitochondrial DAMPs (also known as mitochondrial alarmins) can become pro-inflammatory and initiate innate and adaptive immune responses by activating cell surface and intracellular receptors. Current evidence indicates that uncontrolled and excessive release of mitochondrial DAMPs is associated with severity, has prognosis value in human diseases, and contributes to the dysregulated process observed in numerous inflammatory and autoimmune conditions, as well as in ischemic heart disease and cancer. Herein, we review that the expanding research field of mitochondrial DAMPs in innate immune responses and the current knowledge on the association between mitochondrial DAMPs and human diseases.
Keywords: alarmins; damage-associated molecular pattern; inflammation; mitochondria; pro-inflammatory cytokines; sterile inflammation.
Figures
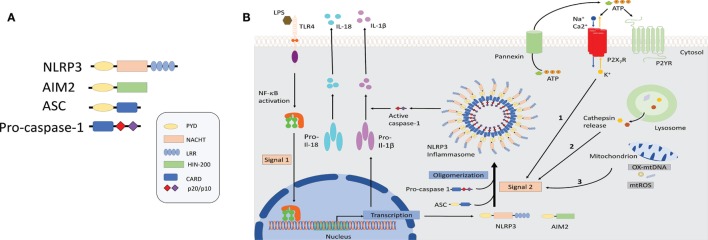
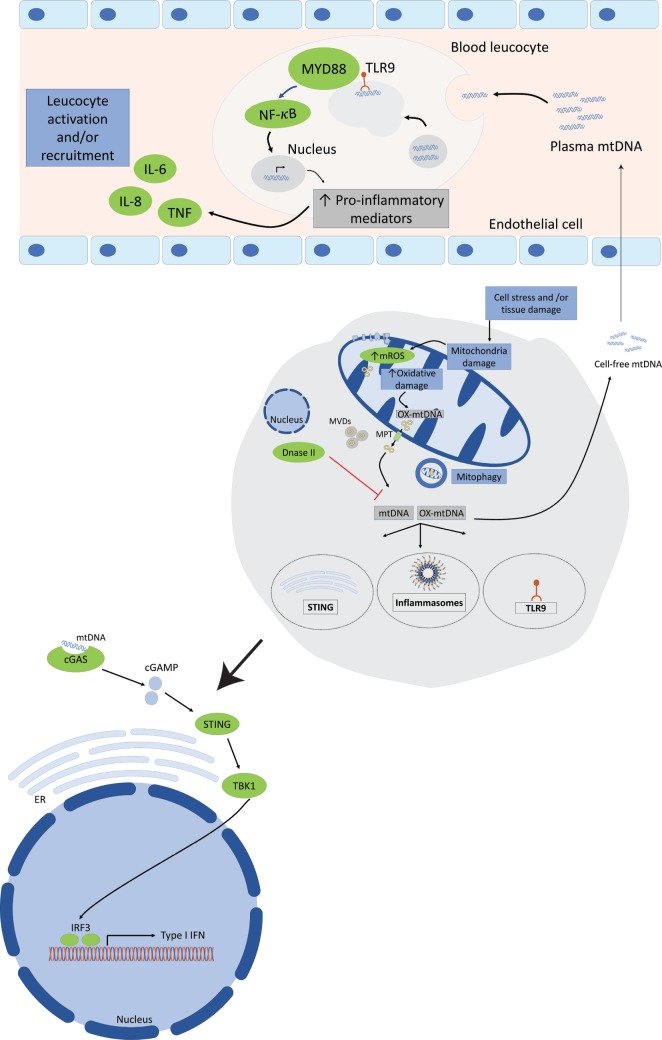
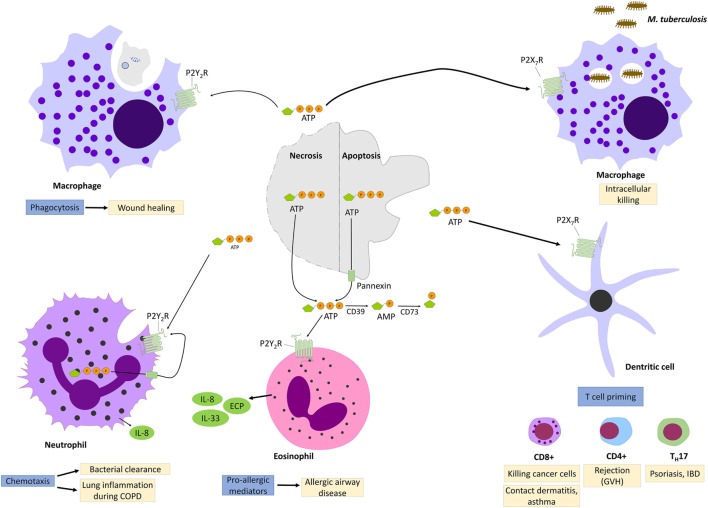
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources