

SAMSA2: a standalone metatranscriptome analysis pipeline
- PMID: 29783945
- PMCID: PMC5963165
- DOI: 10.1186/s12859-018-2189-z
SAMSA2: a standalone metatranscriptome analysis pipeline
Abstract
Background: Complex microbial communities are an area of growing interest in biology. Metatranscriptomics allows researchers to quantify microbial gene expression in an environmental sample via high-throughput sequencing. Metatranscriptomic experiments are computationally intensive because the experiments generate a large volume of sequence data and each sequence must be compared with reference sequences from thousands of organisms.
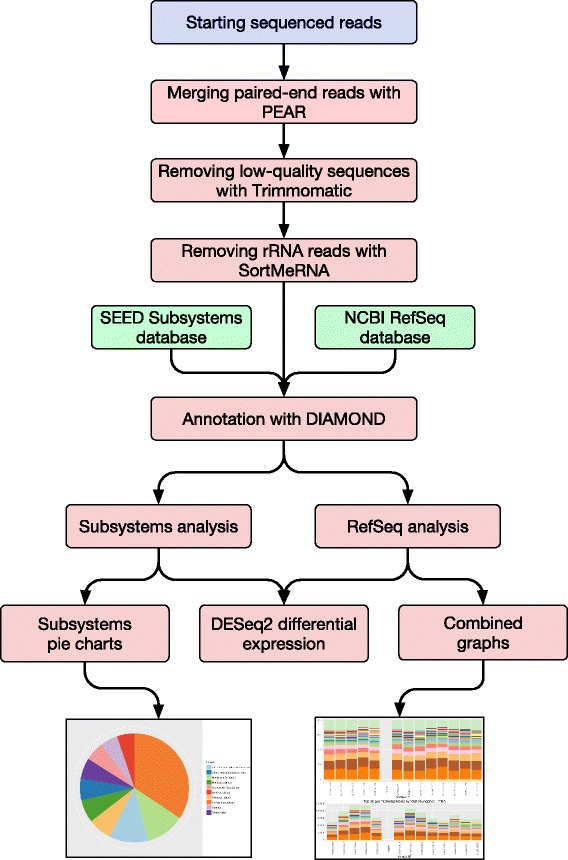
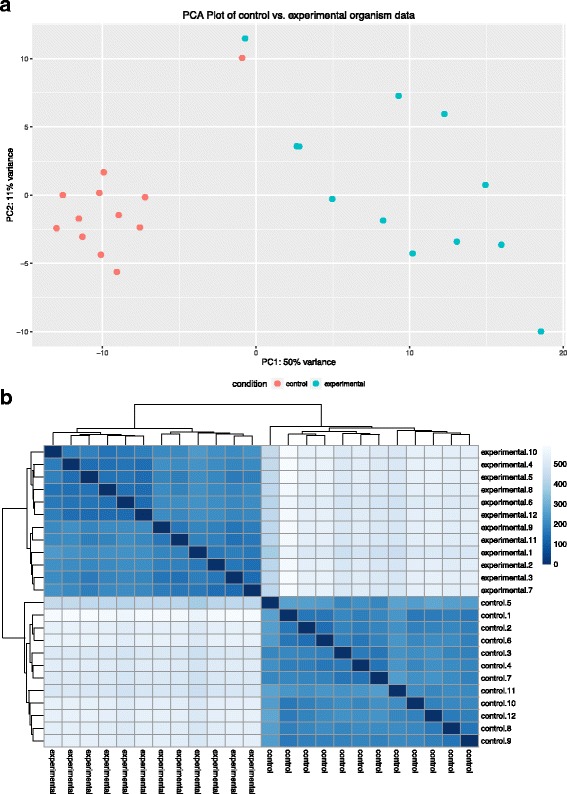
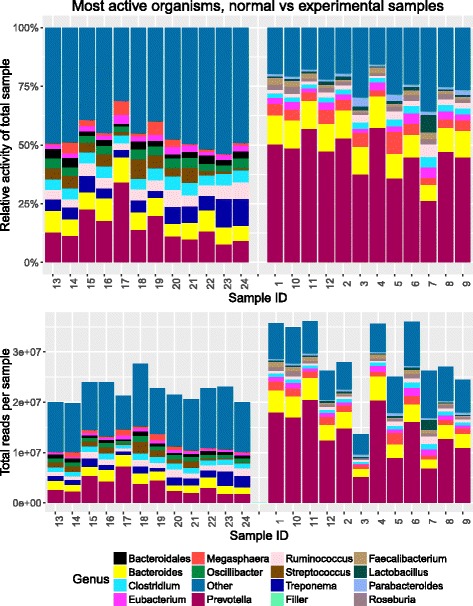
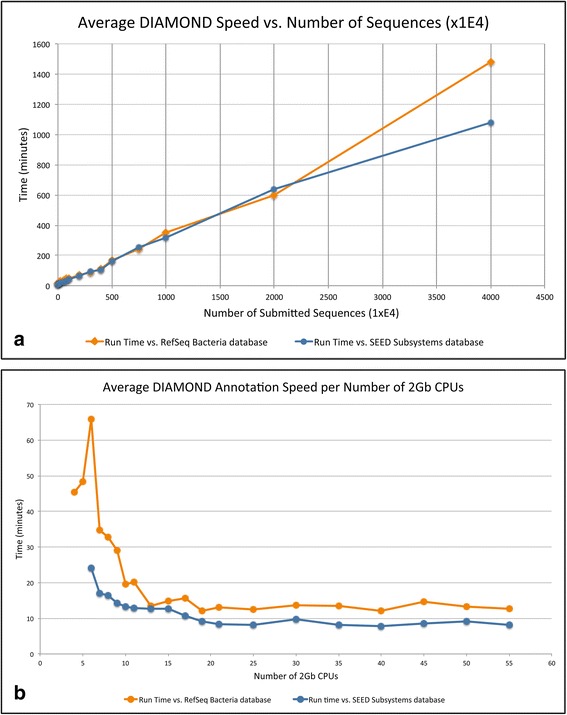
Results: SAMSA2 is an upgrade to the original Simple Annotation of Metatranscriptomes by Sequence Analysis (SAMSA) pipeline that has been redesigned for standalone use on a supercomputing cluster. SAMSA2 is faster due to the use of the DIAMOND aligner, and more flexible and reproducible because it uses local databases. SAMSA2 is available with detailed documentation, and example input and output files along with examples of master scripts for full pipeline execution.
Conclusions: SAMSA2 is a rapid and efficient metatranscriptome pipeline for analyzing large RNA-seq datasets in a supercomputing cluster environment. SAMSA2 provides simplified output that can be examined directly or used for further analyses, and its reference databases may be upgraded, altered or customized to fit the needs of any experiment.
Keywords: Annotation; Bacteria; Bioinformatics; Cluster; Functions; GALAXY; Metagenomics; Metatranscriptome; Metatranscriptomics; Microbiome; Open access; Pipeline; RNA-seq; SAMSA; Software; Tool.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Leimena MM, Ramiro-Garcia J, Davids M, van den Bogert B, Smidt H, Smid EJ, Boekhorst J, Zoetendal EG, Schaap PJ, Kleerebezem M. A comprehensive metatranscriptome analysis pipeline and its validation using human small intestine microbiota datasets. BMC Genomics. 2013;14:530. doi: 10.1186/1471-2164-14-530. - DOI - PMC - PubMed
-
- Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
