

SWI/SNF and RSC cooperate to reposition and evict promoter nucleosomes at highly expressed genes in yeast
- PMID: 29785963
- PMCID: PMC6004078
- DOI: 10.1101/gad.312850.118
SWI/SNF and RSC cooperate to reposition and evict promoter nucleosomes at highly expressed genes in yeast
Abstract
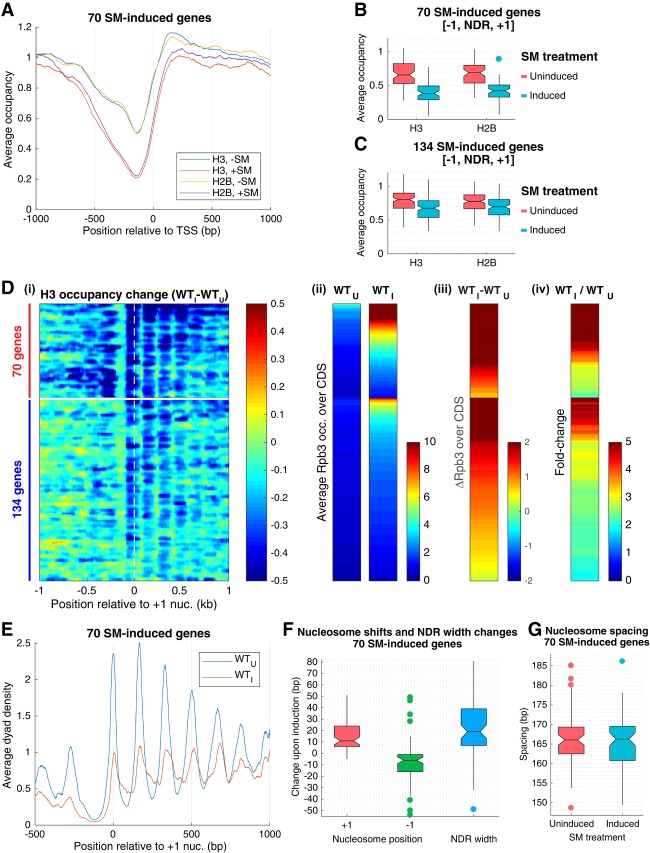
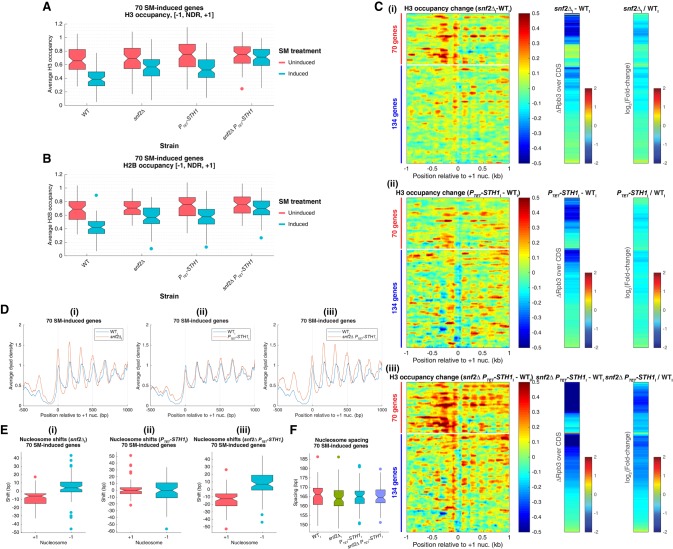
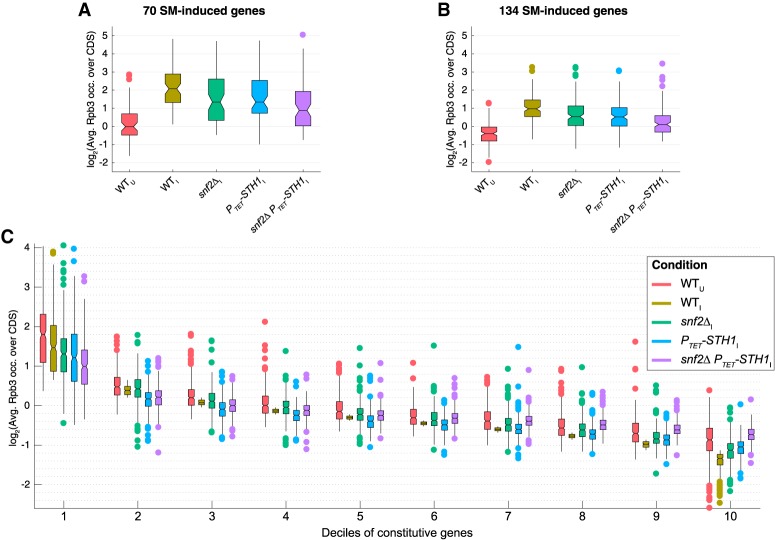
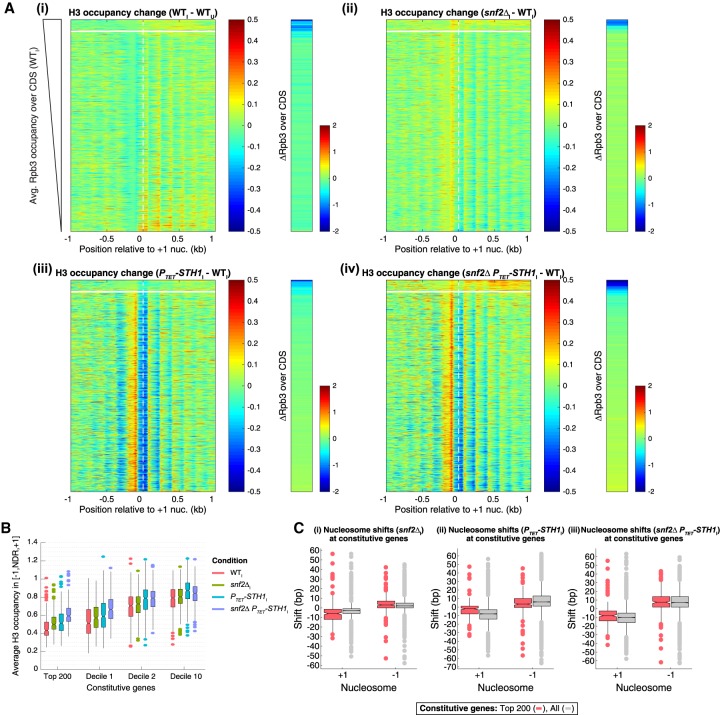
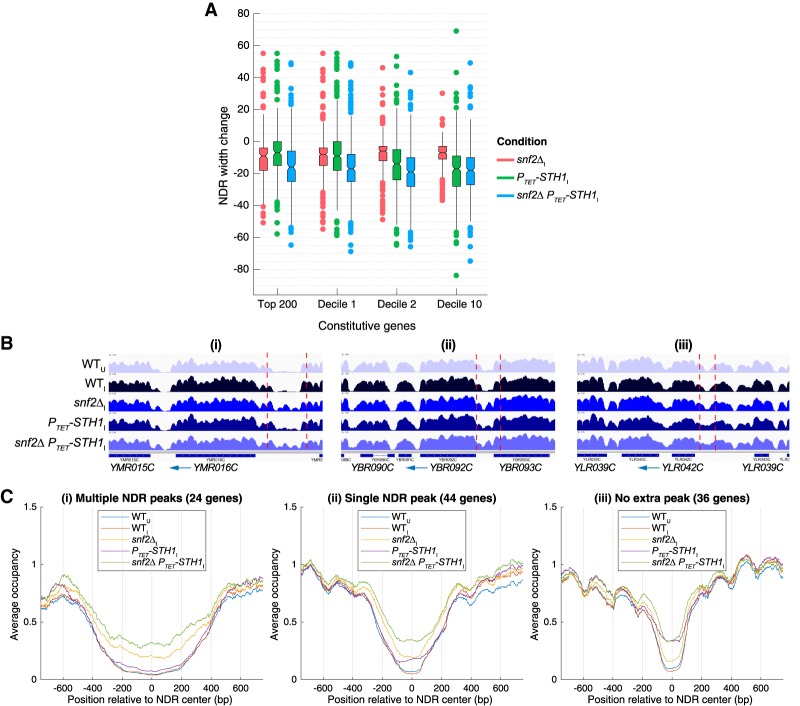
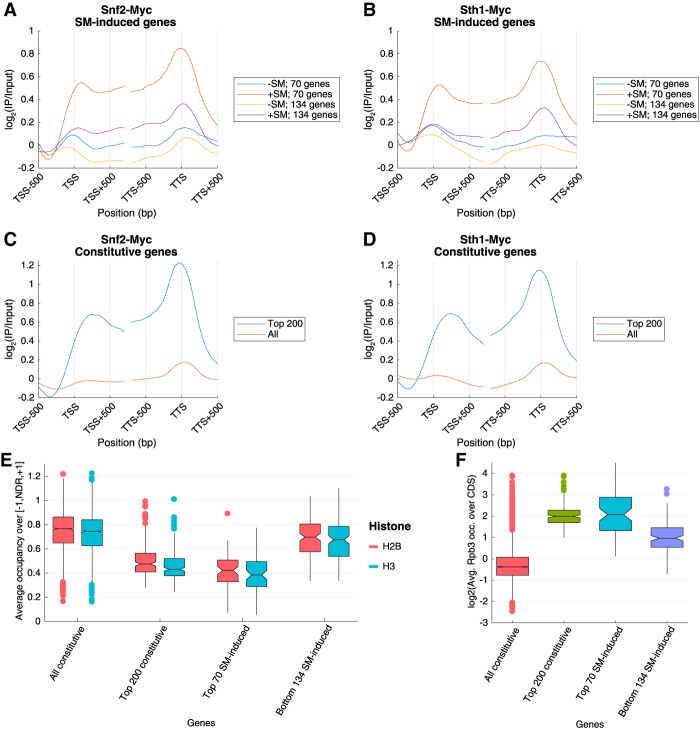
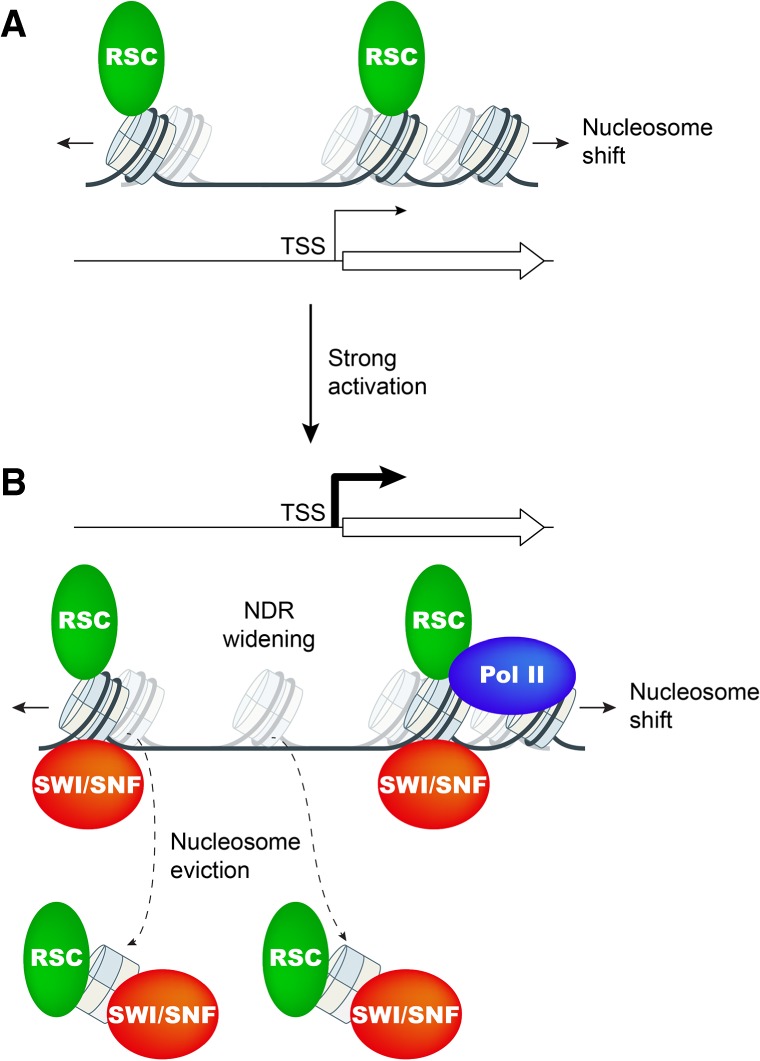
The nucleosome remodeling complex RSC functions throughout the yeast genome to set the positions of -1 and +1 nucleosomes and thereby determines the widths of nucleosome-depleted regions (NDRs). The related complex SWI/SNF participates in nucleosome remodeling/eviction and promoter activation at certain yeast genes, including those activated by transcription factor Gcn4, but did not appear to function broadly in establishing NDRs. By analyzing the large cohort of Gcn4-induced genes in mutants lacking the catalytic subunits of SWI/SNF or RSC, we uncovered cooperation between these remodelers in evicting nucleosomes from different locations in the promoter and repositioning the +1 nucleosome downstream to produce wider NDRs-highly depleted of nucleosomes-during transcriptional activation. SWI/SNF also functions on a par with RSC at the most highly transcribed constitutively expressed genes, suggesting general cooperation by these remodelers for maximal transcription. SWI/SNF and RSC occupancies are greatest at the most highly expressed genes, consistent with their cooperative functions in nucleosome remodeling and transcriptional activation. Thus, SWI/SNF acts comparably with RSC in forming wide nucleosome-free NDRs to achieve high-level transcription but only at the most highly expressed genes exhibiting the greatest SWI/SNF occupancies.
Keywords: RSC; SWI/SNF; nucleosome; transcription; yeast.
© 2018 Rawal et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Barbaric S, Luckenbach T, Schmid A, Blaschke D, Horz W, Korber P. 2007. Redundancy of chromatin remodeling pathways for the induction of the yeast PHO5 promoter in vivo. J Biol Chem 282: 27610–27621. - PubMed
-
- Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. 2003. Nucleosomes unfold completely at a transcriptionally active promoter. Mol Cell 11: 1587–1598. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases